
Abstract
Niemann-Pick type C (NPC) is a rare autosomal recessive disorder characterized by storage of unesterified glycolipids and cholesterol in lysosome. NPC’s clinical presentation is highly heterogeneous, depending on the time of onset. It encompasses visceral, neurological, and/or psychiatric manifestations. As the motor findings are so important and devastating in this disease, there is a lack of description about non-motor symptoms, even though they play important role in quality of life of NPC patients. We described the most common non-motor findings in NPC like cognitive dysfunction, neuroimaging, psychiatric symptoms, sleep disorders, seizures, hearing problems, respiratory and other systemic features, bladder and fecal dysfunction, hypersalivation, and malnutrition. In this review, we highlighted the importance of these undervalued symptoms and their management. Specific measures of all aforementioned clinical features may work as relevant biomarkers in order to evaluate successful therapies in future clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Niemann-Pick type C (NPC) is a rare lysosomal storage disorder caused by mutations in NPC1 (95%) or NPC2 (5%) genes [1]. It is currently conceived as a lipid trafficking disorder. Impaired transport of cholesterol from the late endosomal/lysosomal compartment is a key element of NPC pathogenesis, but other lipids, like sphingolipids, are also involved [2]. In particular, glycosphingolipids (GSLs) are mainly accumulated in neuronal cells. Miglustat is subtract reduction therapy which acts as iminosugar inhibitor of glucosylceramide synthase, which catalyzes the initial step the synthesis of GSL). It is the only drug currently approved for neurological manifestations of NPC in Europe and many countries (but not USA) with focus in the glycolipid storage of NPC pathogenesis. [1,2,3]
NPC’s clinical presentation is highly heterogeneous, depending on the time of onset. It encompasses visceral, neurological, and/or psychiatric manifestations [3]. Typical clinical features include variable degrees of cerebellar ataxia, vertical ophthalmoplegia, and cognitive impairment.
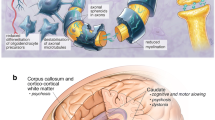
In this review, we provided relevant information of non-motor and extracerebellar manifestations of NPC. Although there are lots of materials about non-motor symptoms in other diseases like Parkinson’s disease (PD) and spinocerebellar ataxias (SCA), these symptoms are underrecognized in NPC. The main non-motor manifestations of NPC described are listed in Table 1 and the most affected areas in NPC are shown in Fig. 1 [2, 4, 5] (Table 1; Fig. 1).

Most affected areas in NPC and their symptom association
Search Strategy
References for this review were identified through searches of PubMed with the search terms “Niemann-Pick type C”, “nonmotor symptoms”, “extracerebellar features”, “Niemann-Pick type C with cognitive decline”, “neuropsychological studies in Niemann-Pick type C”, “neuroimaging in Niemann-Pick type C”, “psychiatric findings in Niemann-Pick type C”, “sleep disorders in Niemann-Pick type C”, “seizures and Niemann-Pick type C”, “systemic features and Niemann-Pick type C”, “autonomic symptoms in Niemann-Pick type C” from the time the disease was first described, in 1961, until march 2020 [6]. Articles also were identified through searches of the authors’ own files. Only articles that were published in English were reviewed. The final reference list was generated based on originality and relevance to the broad scope of this review. The main non-motor symptoms and extra-cerebellar signs in patients with Niemann-Pick type care are described in Table 1.
Cognitive Dysfunction
Cognitive dysfunction is very common in NPC with prevalence ranging from 62 to 100% in previous reports [3, 7]. We analyzed 19 articles about cognitive dysfunction (Table 2).
The earlier neuropsychological papers describe memory, language, spatial-constructional abilities, and executive function impairment even in early stages in a population of NPC patients ranging from 12 to 43 years old. The authors also described severe impairment in verbal fluency and action planning, as well as in expressive vocabulary and drawing skills [8, 9]. A more recent study with 21 patients above 15 years old described executive functions and attention as the most impaired functions [10]. Another study compared NPC (N = 7) and Alzheimer’s disease patients (AD) (N = 15). The authors described relatively preserved verbal memory, but frequent impairment in visual memory, visual construction, executive functions and, in particular, verbal fluency in the NPC group. In the AD group, the more severe neurocognitive deficits were described, primarily featuring verbal and visual memory deficits along with major executive impairment. Delayed verbal memory recall was a particularly strong distinguishing factor between the two groups [11].
Neuropsychological profile in NPC is variable [12]. Mini-Mental State Examination (MMSE) is not a sensitive diagnostic technique for measuring the early cognitive alterations in NPC8, [9, 10] The Trails Making Tests and Grooved Pegboard tests were best suited for patients in the earlier stages of the disease, whereas the Find Similarities test is more appropriate for later stages of the disease, when the patients’ severe lack of fine motor skills preclude the use of the first two tests [10] (Table 3).
Most late onset NPC cases are diagnosed within the third decade. However, recent reports describe initial symptoms at 50 years of age or older, often mimicking common neuropsychiatric illnesses such as parkinsonian disorders, like progressive supranuclear palsy (PSP), or spectrum of frontotemporal dementias (FTD) [3, 13]. Of note, C9orf72, a progranulin gene associated with TAR DNA binding protein-43 (TDP-43), and MAPT, a tau gene both have been linked with early onset neurodegenerative processes (< 40 years of age) in patients with FTD. NPC cognitive deficits also mimic these mutations. Indeed, both NPC and FTD can display similar tau isoform profiles [14]. Hepatosplenomegaly, when present, helps to differentiate NPC from FTD and other etiologies associated with early onset dementia. Abnormal saccadic eye movements (including VSGP) also serve as a strong indicator of possible NPC [14]. As parkinsonian disorders also have a cognitive decline, it is important to distinguish them from NPC. A case series in four PSP patients reported filipin test results consistent with NPC, but no genetic confirmation was established [15].
We found two manuscripts with screening in these dementia patients, one with PSP, PD, and FTD patients without carriers found and the other in 24 PSP and 10 MSA-type C patients found only one MSA as heterozygous to NPC [16, 17]. As occurs in heterozygous Gaucher carriers the development of PD, a review of 19 cases with heterozygous NPC mutations described the diagnosis of neurodegenerative disease with cognitive decline, including levodopa-responsive PD, atypical parkinsonism (PSP, CBD), or dementia with a mean age at onset of about 57 years (range 8–87). [18, 19]
Miglustat is a potential treatment option for cognitive decline [20]. Unfortunately, there is no evidence that cognitive-enhancing drugs such as cholinesterase inhibitors have a beneficial role in these patients. [21].
Neuroimaging
Cerebellar atrophy and white matter changes are the most common neuroimaging findings in NPC. Pontine-to-midbrain ratio seems to be increased in adult patients with NPC when compared with controls. Although the difference between groups was of 14% of the ratio, it was not significant probably due to the small number of ten NPC patients of the study [22]. To analyze NPC progression, global callosal measures were also correlated with duration of illness and symptom score and even with degree of filipin staining, with significant findings in NPC adult patients compared to controls [23]. We found 12 articles about neuroimaging in NPC (Table 4).
A neuroimaging comparative study included six patients with NPC early infantile onset (< 2 years of age), three with late infantile onset (2–6 years), four with juvenile onset (6–15 years), and six with adult onset (> 15 years). The authors described supratentorial atrophy as the leading sign in the infantile groups. Infra and supratentorial atrophy were typical in juvenile and adult forms [24]. White matter changes were found in nearly every patient; with the earlier forms showing progressive worsening through disease duration and the later forms showing prominent white matter changes already early in the disease course [24].
Diffusion tensor imaging (DTI) analyses showed decreased fractional anisotropy in NPC patients compared with controls, especially in corpus callosum, internal capsule, corona radiate, and cingulate gyrus, with an early but transient improvement of DTI metrics after miglustat treatment [25, 26] A study with a combination of optimized voxel-based morphometry of T1-weighted images and tract-based spatial statistics of diffusion tensor images cortical thickness analyses showed an atrophy pattern in thalamus, hippocampus, caudate nucleus, and cerebellum [27].
Functional imaging with 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) and SPECT has highlighted frontal and temporal alteration, even at the beginning of disease [28]. Proton magnetic resonance spectroscopic imaging (H-MRSI) studies have shown a decreased N-acetyl aspartate/creatine ratio in frontal and parietal cortices, centrum semiovale, and caudate nucleus, with significant correlations between clinical staging scale scores and H-MRSI abnormalities [29, 30] In an amyloid PET report of two adult sisters, cortical amyloid deposition was found in frontal areas [31].
Although these findings and specific neuroimaging features are lacking in NPC, providing little aid in the clinical setting and diagnostic workup [5, 32], NPC shows pathophysiological findings as increased levels of brain tau protein, amyloid deposition, neurofibrillary tangles (NFTs), neuronal degeneration, neuroaxonal dystrophy, demyelination, and the influence of apolipoprotein E ε4 genotype which have a greater relation to cognitive symptoms [32, 33].
Psychiatric Symptoms
Psychiatric symptoms are commonly reported in NPC. Differential diagnosis of NPC versus primary psychiatric conditions and other organic causes can be very challenging [1, 14, 34] Psychosis in NPC is usually reported with persecutory delusions, hallucinations, ideas of reference, and behavioral disorganization [14, 18, 35]. Other psychiatric disturbances include bipolar disorder, depression, anxiety, obsessive-compulsive disorder more common among juvenile and adult forms [35]. In earlier forms, manifestations like autistic, intellectual disability, impaired impulse control, mood, and anxiety can occur, besides psychosis [34].
Not uncommonly, patients may present psychosis as the first clinical manifestation of the disease [34, 36]. Some red flag symptoms to remember NPC are acute confusional state, preponderance of visual over auditory hallucinations, early or acute onset, and a fluctuating course of symptoms with refractoriness [14, 35]. Pseudobulbar affect associated to psychosis later in disease is also reported in NPC patients [7]. Importantly, in a comprehensive study of 13 unrelated adult patients diagnosed in France over 20 years and the analysis of the 55 other cases published since 1969 until 2005, the authors concluded that psychiatric features rarely constituted a late complication, with only two of their patients presenting psychiatric troubles after an initial motor presentation [37].
Table 5 describes the main difference between primary psychiatric symptoms and NPC psychiatric symptoms. We analyzed 15 articles about psychiatric findings in NPC disease that are enrolled here (Table 6).
Previous studies have reported psychiatric manifestations in 25–45% of NPC patients, with psychotic and mood symptoms as the most frequent [4, 38, 39]. A multicenter, prospective, observational study found psychiatric symptoms in 34% (94/280) of NPC patients, with the most frequent being psychotic (n = 19; 43%), followed by mood (n = 17; 39%) and impaired impulse control (n = 8; 18%) [34]. Another retrospective survey of psychiatric presentations among French adult NPC patients reported psychotic symptoms in 55%, and half had a psychiatric diagnosis: schizophrenia in 27% and depression in 23% [40].
A systematic review analysis of psychiatric symptoms in 58 NPC, mainly juvenile and adult forms, reported psychotic symptoms in 62% overall, and behavior and mood-related manifestations in 52% and 38%, respectively [34]. Similarly to many diseases associated with organic psychiatric symptoms, NPC patients who initially present in psychiatric practice may remain improperly diagnosed for years due to heterogeneous clinical features or the lack of other symptoms besides psychiatric in first years [41]. Limited knowledge of NPC among clinical physicians is a possible underlying explanation to this problem [41, 42] Indeed, 6 years to even decades delay on diagnosis is described among these psychiatric patients [20, 38, 41]
Schizophrenia could be explained by a profound disruption of large-scale prefrontal-temporal interactions, not only in physiology and functional anatomy, but at the level of cognitive and sensorimotor functioning [43]. The physiopathology of psychiatric symptoms in NPC is not entirely known, but looks like there are alterations in connectivity of synapse, myelinated axons, and disrupted function in more distributed cortico-subcortical networks as frontotemporal myelination, dorsolateral prefrontal cortex (DLPFC), and temporoparietal-occipital association areas impairing frontotemporal connectivity and frontal-subcortical connectivity as described in schizophrenia [26, 43].
Limbic structures in the medial temporal lobe have also been intimately implicated in the pathophysiology of schizophrenia and have been seen as central to the neuropathology of NPC [44]. The insula plays a key role in processing of sensory inputs and with visual–tactile and auditory–visual integration and shows reduced surface area in schizophrenia with abnormal activation apparent on fMRI (functional MRI) in auditory hallucinations [44]. This is an area of significant NFT accumulation and regional volume loss in adult NPC patients [44]. Also, as endogenously synthesized cholesterol is necessary for axonal membrane maintenance and repair, white matter tracts are severely affected, with the corpus callosum, showing the most striking axonal loss which could lead to psychosis [20, 45]
Due to its prevalence, a screen study with consecutive patients aged ≥ 18 years undergoing medical care in psychiatric or neurological reference centers across the European Union and USA, “ZOOM”, systematically tested for NPC gene mutations in 250 patients with neurological and psychiatric symptoms, fulfilling one of these criteria: psychosis combined with ≥ 1 pre-defined neurological or visceral symptom/syndrome; early-onset progressive cognitive declined combined with ≥ 1 pre-defined neurological or visceral symptom/syndrome; early-onset progressive cognitive decline combined with psychosis [39]. The authors identified three (1.2%) new NPC cases, and 12 (4.8%) more patients who were classified as “NPC uncertain”, showing a much higher frequency of NPC in this population [39].
Oxysterols species cholestane-3β, 5α, 6β-triol (C-triol), nowadays, is the most used biomarker for NPC testing with clinical sensitivity of even 100% and specificity of 93.4% [5, 46]. Neurofilament light chain (NFL) is a biomarker of neuronal injury that has shown great promise in differentiating NPC patients from primary psychosis, being more feasible and cost-effective as screening tool for NPC before testing oxysterols [46].
Psychosis in NPC usually responds to antipsychotic medications, preferably atypical ones. However, some patients are resistant to treatment or even show (paradoxical) worsening with the initiation of drug therapy (a useful diagnostic red flag in unidentified NPC) [20]. Depression typically responds well to SSRIs, and in some patients, when effectively treated, this leads to improvements not only in their mood but also in their cognition and neurological disease. Bipolar disorder in NPC has responded to mood stabilizers such as sodium valproate and catatonia has been treated successfully with ECT [20].
Sleep Disorders
There are few publications investigating the association between NPC and sleep disorders, besides cataplexy [7, 47, 48]. Only Vankova and Rangel performed polysomnography (PSG) with multiple sleep latency test (MSLT), in five and four NPC patients, respectively, with clinical evaluation by interview and/or sleep scales made only in the last one. [7, 47] Rangel et al. (2019) demonstrated other sleep disorders, besides cataplexy, in this population like chronic insomnia (62.5%), probable and confirmed obstructive sleep apnea (OSA) (62.5%), symptoms of REM sleep behavior disorder (RBD) (25%), and restless legs syndrome (RLS) (25%).
Excessive sleepiness also has been mentioned by some authors like Rangel et al. who described it as the first symptom of the disease along with sleepwalking in a patient and Nevsimalova et al. who also described an excessive sleepiness in one of their cases [7, 48]. Sleep-wake circadian disorder, narcolepsy, or OSA were reported in other paper [38]. Rangel et al. hypothesized a relation between chronic insomnia and duration of disease, since their patients with this complaint all had more than 10 years of disease, like a patient described by Nevsimalova [7, 48].
Kawazoe et al. (2018) described an infantile-onset NPC case with minimal neurological symptoms and excessive daytime sleepiness at the age of 17 besides sleep paralysis, no polysomnography was made. Her mother was accompanied with narcolepsy and was heterozygous for the mutation of c.160_161insG (p.D54GfsX4) on NPC1 gene found in the patient [49]. Despite her mother be heterozygous NPC carrier, there is a previous report of alterations in NPC carriers with foam cells in bone marrow, splenomegaly, and parkinsonian syndromes [50]. Therefore, the production of hypocretin at the lateral hypothalamus may be affected in NPC, which could explain excessive sleepiness [47]. Unfortunately, we could not find pathological reports in human heterozygous NPC to better understand their physiopathology.
Gelastic cataplexy is a finding more specific than prevalent between NPC patients, with classically description among these patients [48]. It occurs mostly in late infantile and juvenile forms, with prevalence of 20% NPC children [51]. So, children with isolated gelastic cataplexy should undergo NPC testing [5, 52] It is a sudden and brief loss of muscle tone evoked by a strong emotional (humorous) stimulus. Minor or partial attacks are common and can be difficult to recognize even for close family members. Slight drooping of the head, slurred speech, or dropping an item from the hand, for example, may be the only observable manifestation of a cataplectic event [52, 53]. NPC is associated with increased cholinergic activity and decreased monoaminergic activity in the upper pontine tegmentum, both essential in the transition from non-REM to REM sleep. These findings could explain this symptom [7, 48] Although it has its specificity, cataplexy may also be a component of other rare genetic syndromes, like Norrie disease, Coffin–Lowry syndrome and autosomal dominant cerebellar ataxia, deafness and narcolepsy (ADCA-DN) [52].
Polysomnography seems to show disorganized sleep with lower-than-expected sleep efficiency, total sleep time, and total amount of REM sleep compared to normal individuals of the same age and gender [7, 47] Excessive fragmentary myoclonus has been described in patients with this disease [7, 47] NPC patients also appear to have poor quality sleep with presence of sigma activity, atypical forms of sleep simples and K complexes and alphadelta pattern in electroencephalogram (EEG) [7, 47]. MSLT seems to be abnormal in some NPC patients with lower mean sleep latency and early REM sleep onset (SOREM) [7, 47].
There are theories that occur a gradual and progressive involvement of structures related to the sleep-wake cycle in the brainstem and diencephalon as NPC progresses. Neuronal death becomes overt, predominantly affecting certain regions, like cerebellum Purkinje cells. Indeed, the involvement of the cerebellum in sleep control was reported previously, with the connection of sleep-wake network by cerebellum output neurons. As a result, patients with ataxia could present a variety of sleep disorders [7, 54].
There are no studies about Miglustat effect on sleep. In chronic insomnia, sleep hygiene and hypnotic drugs, like zolpidem, seems to have good response [7]. Patients with OSA could be treated with continuous positive airway pressure device (CPAP) or positional therapy, depending on the severity of respiratory events and the worse of them during supine position [5, 7] Other types of OSA treatment have not been reported in NPC patients. For excessive sleepiness caused by the disease, methylphenidate and naps seem to have good response [7]. In clinical practice, the most used drugs in cataplexy are venlafaxine (doses 75–225 mg daily) or clomipramine (25–100 mg daily). Other antidepressants such as fluoxetine or other specific serotonin-reuptake inhibitors can also suppress cataplexy in some people [51]. Sleep-wake cycle disorder and RBD can be treated with melatonin [5].
Epilepsy
Epilepsy is a common unspecific finding in NPC disease. Its epileptogenesis is yet to be explained. Xu et al. have demonstrated an important defect on glutamatergic and gamma aminobutyric acid-ergic exocytosis, caused in part by a delay for replacing vesicles with new competent ones [55]. This turnover delay was longer in inhibitory pre-synaptic level, gamma aminobutyric acid-ergic exocytosis, than in excitatory ones, which could explain the seizures on the disease [55]. Other possible published mechanism on development of NPC neuroinflammation include reduced intercellular communication via gap junction and increased hemichannel activity represented by the findings of newborn NPC1 astrocytes on hippocampal slices with high hemichannel activity [56]. These hemichannels mediate the uptake or release of ions and small molecules such as Ca2+ and ATP, playing a role in the development of neuroinflammation in NPC disease. NPC1 astrocytes also showed more intracellular Ca2+ signal oscillations mediated by functional connexin in 43 hemichannels and P2Y1 receptors. Further investigations are required to elucidate the mechanism of neurodegeneration and epileptogenesis [56]. We analyzed 14 articles about seizures occurrence in NPC disease that are enrolled here (Table 7).
The international disease registry of 163 NPC patients described seizures in 90 (33%) patients. Seizures and cataplexy were more common among late infantile and juvenile-onset cases (37–57%) than early infantile (17%) and adolescent/adult onset (6–9%) [3, 5, 20].
In general, seizures are more commonly noted as the disease progresses [41]. When occurs at diagnosis of disease, its prevalence is higher in patients diagnosed from 10 to 20 years of age [21]. When occurs later, it may reflect a more severe or advanced state [57].
NPC patients can experience any type of seizure like focal or generalized (absence, myoclonic, tonic-clonic), without predominance of any kind. They vary markedly in intensity and frequency [38]. Refractory epilepsy has been reported in several NPC cases, all of them with previous neurological symptoms [38]. There are no reported cases with refractory epilepsy as the sole presenting manifestation of NPC [58]. Patients may have seizure-like episodes including arrhythmic cortical myoclonus and stimulus-sensitive myoclonus [58].
Refractory seizures are rarely described in all forms of disease. A previous report of late-infant NPC patient with refractory seizures described a successful treatment with levetiracetam, valproic acid, and nitrazepam [59]. One NPC patient presented refractory seizures at the age of 22 years with a mix of gelastic cataplexy and myoclonic seizures occurring up to 30 times per day [41]. Another case in adult form, with psychosis on disease onset, evolved with several different types of seizures [41].
Electroencephalographic (EEG) features consist of nonspecific diffuse slowing of background activity and interictal discharges without specific pattern [60, 61]. Rangel et al. described presence of alphadelta pattern in three patients and disorganization of background electroencephalographic activity in two patients [7]. EEG is normal in gelastic cataplexy and shows high-frequency oscillations in case of cortical myoclonus [58]. Brain imaging changes pertaining to specific seizure type in NPC have not been described [60].
Carbamazepine, oxcarbamazepine, and vigabatrin should be avoided in NPC seizures as they could promote myoclonus. Phenytoin should not be used in order to avoid possible cerebellar adverse effects [5]. Anti-epileptic drugs applied to the NPC patients include sodium valproate, lamotrigine, and levatiracetam [58]. Information regarding the effect of miglustat on seizures is limited and conflicting [62, 63].
Systemic Features
Hepatosplenomegaly is a common feature of the earlier forms of NPC, being less frequently among juvenile and adult onset forms [5, 21]. Historical or current isolated unexplained splenomegaly, with or without hepatomegaly, appears in 58% of NPC patients, being the strongest visceral indicator of the disease [38]. A group reported splenomegaly (with or without hepatomegaly) on abdominal ultrasound in closer to 90% of patients, regardless patient’s age [37]. Ultrasound should always be made in NPC suspicion patients, since mild splenomegaly may only be detected by abdominal imaging [64, 65]. Bonnot et al. even incorporated abdominal ultrasound to their diagnostic ‘work-up’ algorithm for IEMs causing a schizophrenia-like illness [35].
Interestingly, regardless of the patient’s age, visceral disease (when present) always precedes neuropsychiatric features, often by years or even decades. There are cases of patients with pediatric liver disease who only develop neuropsychiatric features many decades later in adulthood. [20, 41]
When present in combination with other neurological and/or psychiatric symptoms, including vertical supranuclear gaze palsy (VSGP), ataxia and schizophrenia-like symptoms, isolated splenomegaly becomes highly suggestive of NPC [38]. Isolated unexplained splenomegaly should lead to the inclusion of NPC in the differential diagnosis, and hence trigger a search for other symptoms. Splenomegaly in NPC ranges from slight to tremendous enlargement. Importantly, the degree of splenomegaly does not correlate with disease severity or illness stage. Absence of splenomegaly should not lead to the exclusion of NPC, not even in the earlier forms [21]. We analyzed 10 articles about systemic features in NPC disease (Table 8).
Hepatomegaly is less frequently observed in adult patients with NPC, with non-specific presentation. It generally appears at the same age of splenomegaly [38]. Notably, the degree of hepatomegaly and splenomegaly are not related, and the first one does not appear to resolve spontaneously, unlike the second [1]. Miglustat does not appear to have any effect on visceral symptoms. Patients with hepatosplenomegaly do not show any improvement in liver or spleen volume [62].
In a review of 164 NPC patients, hydrops fetalis and siblings with fetal ascites were not commonly reported at diagnosis, with a prevalence below 10% in all age groups [21]. Signs of perinatal liver involvement in NPC cases range from transient conjugated hyperbilirubinemias to severe cholestatic hepatopathy. Conjugated bilirubin levels and speed of symptom resolution are non-specific in NPC [65]. Since this condition does not require phototherapy (unlike unconjugated jaundice), its symptoms may often not be recalled by parents and hence may be missed when obtaining medical history [65].
Lung disease can occur in 13% of NPC patients, but is more associated with NPC2 and usually with severe types of disease [66, 67]. In NPC2, the clinical picture can be similar to chronic lung disease of the newborn in the absence of a history to support it. Chest-computed tomography may show classical interstitial lung disease [21]. These features have been poorly described in the literature but are often reported by experts. There is no specific therapy for pulmonary manifestations. Clinical report of 16-month-old boy homozygous for NPC2 showed pulmonary response after bone marrow transplantation [68]. It is commonly treated with symptomatic medications like aggressive bronchodilation and, in some cases, chest physical therapy [38].
Mild thrombocytopenia in early infantile NPC has been reported. Usually, patients with classical foamy cells are the most severely affected and present large splenomegaly, low platelet counts, bone infiltrates, and a widespread presentation, including neurological manifestations [21, 38]. Another findings described in adult case reports were insulin-dependent diabetes mellitus, thalassemia, glomerular nephropathy, and bilateral carpal tunnel syndrome, which could be coincidental since there are no other reports [37]. Some papers show electromyographic signs of myelinic neuropathy symptomatic and asymptomatic before the use of miglustat [63].
Hearing Problems
There is paucity of studies describing auditory system symptoms in patients with NPC. We found a total of 11 articles containing information about this feature. High frequency of sensorineural hearing loss was reported in NPC in different intensity [1, 21]. It affects about 20% to 50% of patients and appears to be more frequent in adults and with progressive manner [65]. It is believed that cholesterol, whose trafficking is impaired in NPC due to lysosomal storage, plays a key role in auditory physiology, at inner ear. Hearing ability can be tested by audiograms or auditory brainstem responses on evoked potential. [69, 70]
Detailed audiovestibular and swallowing evaluation was only described in two studies. The first one included 16 NPC patients. The authors reported a high frequency of Auditory Brain stem response (ABR) abnormality in at least one ear (N = 12; 75%). Of these, four patients had normal hearing and three of them had peripheral hearing loss [69]. The second one encompassed 31 NPC patients who underwent behavioral evaluation. Twenty-three (74%) presenting significant hearing loss [71]. Data from NPC patients reveal high frequency hearing loss and mixed type hearing loss which are more common in patients with late infantile onset form [67]. Sporadic descriptions of high frequency hearing loss, acoustic reflex abnormalities, and auditory brainstem response disturbance suggest possible widespread auditory dysfunction. [4, 60, 71] Another study with 13 adult NPC forms identified 3 patients with hypoacusia. [37]
Auditory manifestations may have been underreported given the inability of many affected individuals to self-report hearing difficulties due to cognitive decline. Due to its progressive form, early hearing stimulation is important in these patients [72]. Animal studies have shown that the cholesterol-chelating agent 2-hydroxypropyl-β-cyclodextrin, a promising experimental therapy for NPC, may have deleterious effects on hearing impairment, emphasizing the need for auditory testing during this treatment [73].
Autonomic Symptoms
There are only review articles describing autonomic symptoms in NPC with no specific article on this matter [21]. Although it lacks literature about it, bladder dysfunction is a common finding in these patients [4]. Stampfer et al. (2013) described urine and feces incontinence in more than 40% of 42 NPC patients from all the forms. The onset of these problems was later, after 10 years of progression. [57]
A consensus clinical in NPC was made in 2018 approaching fecal impaction and incontinence besides bladder dysfunction, with indication of active search for symptoms suggestive of neurogenic bladder (recurrent urinary tract infection, nocturia, incomplete evacuation, dribbling) [5]. The positive cases should be referred for urologic evaluation [5]. Constipation can also occur. To avoid fecal impaction consider modifying diet and lifestyle to optimize stool consistency. If required, consider appropriate laxatives to optimize gut transit and stool consistency [5].
Hypersalivation/drooling is a common finding in these patients and can be a distressing manifestation [4, 5]. It should be treated with established interventions like oral atropine, parotid/submandibular injections of botulinum toxin, hyoscine patches, or glycopyrronium bromide [38].
Gastrointestinal disturbances such as diarrhea are often evidenced in NPC as an adverse effect of miglustat that tend to decrease over time. Diarrhea can be managed with anti-propulsive agents such as loperamide, and bowel-monitoring programs can be instituted to prevent constipation in affected patients [74].
Transient loss of consciousness such as syncope, vasovagal syncope, or arrhythmias can occur in NPC mimicking seizures [58]. Apparently, there are no reports of sexual dysfunction in NPC patients.
Nutritional Aspects
Malnutrition is a common feature of the disease. Patients progress with weight loss [38]. The affected areas in the disease, like brainstem, or the cortical frontal lobe (areas responsible initiating the swallowing action) could explain this weight loss due to motor dysfunction [5, 38]. Movement disorders, like dystonia, could also explain the energy spoiling and weight loss [75].
Besides the NPC physiopathology, Miglustat therapy is another explanation to body weight reductions possibly related to carbohydrate malabsorption, which may contribute to a negative caloric balance and increased risk of diarrhea [74]. It inhibits the activity of intestinal disaccharidases and food osmolarity, causing diarrhea (17/20; 85%), flatulence (14/20; 70%), weight loss (13/20; 65%), and abdominal pain (10/20; 50%) [74]. Nevertheless, the incidence of diarrhea, flatulence, abdominal pain, vomiting decreased over time after they started using Miglustat [76].
Attention should always be paid to hydration and weight control, with food supplementation required occasionally [73]. In long term, selection of foods could be based on a diet that is very low in carbohydrates, such as the modified Atkins diet, or on a diet that specifically avoids large amounts of critical carbohydrates (such as sucrose, maltose, and starch) [77]. Consideration of the patient’s nutritional status, swallow safety, and toileting/ bowel function, as well as their mobility and safety, is important, with a multidisciplinary team involvement [78].
Discussion
NPC is a rare neurovisceral disease characterized by progressive neurodegeneration with heterogenous clinical presentation accordingly to age of onset [2]. The identification of a broader clinical spectrum of the disease is crucial to better understand and to measure disease progression for future clinical trials.
Executive dysfunction is the main finding in cognitive evaluation. Furthermore, cognitive screening tools are necessary for clinical evaluation in every stage of the disease. Trail Making and Grooved Pegboard tests seems to be the best suited tests to be used in the earlier stages of the disease as well as Find Similarities test for later stages [8, 10]. Neuroimaging also plays a role in excluding other causes in the diagnostic workup [32]. Moreover, global callosal measures, morphometry of T1-weighted images, tract-based spatial statistics of cortical thickness in diffusion tensor images, and spectroscopy findings may help clinical measures of disease progression [23, 25,26,27,28,29,30].
Psychosis in NPC occurs mainly in juvenile and adult forms. The main red flag symptoms that alert NPC on psychosis differential diagnosis are acute confusion state, preponderance of visual over auditory hallucinations, early or acute onset of symptoms, and a fluctuating course of symptoms with refractoriness besides motor symptoms [14, 34]. Based on what we discussed, we suggest some study to evaluate the prevalence of organic conditions (such as NPC) in patients with refractory psychosis. To improve the detection and monitoring of psychotic symptoms, researchers can conduct a meticulous clinical interview and use scales such as the Brief Psychiatric Rating Scale (BPRS), Positive and Negative Syndrome Scale (PANSS), and several versions of the Neuropsychiatric Inventory (NPI) [79,80,81,82].
Patients with ataxia could present a variety of sleep disorders [54]. As described before mainly cataplexy, but also chronic insomnia, OSA, RBD, and RLS have been described in NPC, making sleep evaluation essential to these patients [7, 48]. To evaluate sleep disorder, we could perform a detailed clinical evaluation and, in specific cases, polysomnography and multiple sleep latency test. Besides that, sleep scales could assess sleep disorder’s severity and help the patients’ follow-up, such as Epworth Sleep Scale, Pittsburgh Sleep Quality Index, and International Restless Legs Syndrome Rating Scale, which could also be used to evaluate NPC response in trials [7, 83,84,85].
NPC patients can experience a variety of seizures’ types, like focal or generalized. Seizures and cataplexy were more common among late infantile and juvenile-onset presentation and lead to NPC diagnosis, mostly cataplexy for its especificity [3, 5, 20]. When seizures occur later in the course of the disease, it may reflect a more severe or advanced state [57]. Whenever seizures are suspected, a electroencephalography should be done in order to confirm and characterize the epilepsy disorder. If the doubt about the diagnosis of ictal events persists, the patient could make a more extensive electroencephalography monitoring with synchronized video. National Hospital Seizure Severity Scale (NHSSS) and Seizure Severity Questionnaire (SSQ), which already has a Portuguese version, are alternatives to evaluate seizures and their impact on these patients. [86]
As other neurometabolic disorders, some potential disease-modifiable treatments are available for NPC. Miglustat could reduce the toxic accumulation of GM2 and GM3 gangliosides, which has been shown to improve/stabilize several neurological illness markers in NPC [44]. A clinical observation study with nine adult NPC patients in treatment showed slower cerebellar gray and white matter losses, bilateral thalamic volume, and right caudate volume than untreated NPC patients [87]. In another study, using only Miglustat treatment, two patients showed improvement on their psychosis symptoms [88]. One pediatric patient who presented depression symptoms alongside typical neurological features showed improvement on their mood symptoms after 12 months of treatment with Miglustat [89]. Patterson et al. showed better MMSE scores over the course of 12 months of treatment with Miglustat in 20 patients over the age of 12 compared to nine patients that received standard care [90]. During a therapeutic trial of Miglustat, Hinton et al. described better MMSE scores in 12 out of 14 NPC patients who met the criteria for dementia [8].
As other neurometabolic disorders, some potential disease-modifiable treatments are available for NPC. Intrathecal 2-hydroxypropyl-β-cyclodextrin (2HPßCD) has been shown to markedly improve neuronal cholesterol homeostasis with a promise in slowing disease progression in humans [44, 91]. 2HPßCD is an FDA approved excipient used to dissolve intravenous steroids [92]. Although not yet approved, a current phase IIb/III international trial is currently underway in adult and pediatric patients with NPC (https://clinicaltrials.gov/ct2/show/NCT02534844) [44].
There have been no reports of psychiatric symptomatology in the NPC disease responding to 2HPßCD up to now [44]. Phase I–II trial data showed stable or improved scores in 3 juvenile NPC patients using standardized IQ measures and computerized cognitive tasks treated with 2HPßCD but with hearing loss as adverse effect in all of them [92]. Finally, a review of 16 early published cases of 2HPßCD treated NPC patients, with 13 late-infantile NPC and 4 juvenile forms, suggested that cognition stabilizes or improves in most of these patients. Unfortunately, there was hearing loss in almost a quarter of the cases (four patients, two late infantile, and two juvenile forms) [93]. Animal studies have also shown that 2HPßCD may have deleterious effects on hearing impairment, emphasizing the need for auditory testing during treatment [73].
The remarkable advance of the study of NPC disorder may provide new potential interventions. Of note, several approaches to the metabolic pathway of sphingolipids have been attempted such as histone deacetylase inhibitors and arimoclomol, a heat shock protein (Hsp) activator [94, 95]. Acetylleucine, an acetylated derivative of a natural amino acid, was already used in cerebellar patients with beneficial effects [96]. Observation study with 12 NPC patients (3 late-infantile, 8 juvenile and 1 adult form) demonstrated improvement on balance, coordination and patient’s quality of life, comparing 1 month with acetylleucine and 1 month after washout, with no significant side effects [96]. Finally, there is ongoing trial, prospective, randomized, double-blind, phase II/III, placebo controlled use of arimoclomol as a potential drug for NPC treatment, through the increase in heat shock protein response physiopathology (NCT02612129) [97].
Conclusion
This study highlighted the high frequency of non-motor symptoms in patients with NPC: cognitive dysfunction, psychiatric symptoms, sleep disorders, seizures, hearing problems, respiratory dysfunction, bladder dysfunction, hypersalivation, and malnutrition. Many of these symptoms and signs are potentially treatable and deserve attention, whose treatment may improve patient’s quality of life. Nevertheless, these symptoms are yet undervalued when compared to motor symptoms of disease. Specific measures of all aforementioned clinical features may work as relevant biomarkers in order to evaluate successful therapies in future clinical trials.
References
Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16.
Vanier MT. Complex lipid trafficking in Niemann-Pick disease type C. J Inherit Metab Dis. 2015;38(1):187–99.
Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013;8(12):1750–172.
Garver WS, Francis GA, Jelinek D, Shepherd G, Flynn J, Castro G, et al. The National Niemann–Pick C1 disease database: report of clinical features and health problems. Am J Med Genet A. 2007;143A(11):1204–11.
Geberhiwot, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018;13:50.
Crocker AC. The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J Neurochem. 1961;7:69–80.
Rangel DM, Sobreira-Neto MA, Nepomuceno CR, Marques ER, Braga-Neto P. Sleep disorders in Niemann-Pick disease type C, beyond cataplexy. Sleep Med. 2019;57:122–7.
Hinton V, Vecchio D, Prady H, Wraith E, Patterson M. The cognitive phenotype of type C disease: neuropsychological characteristics of patients at baseline in a clinical trial with oral miglustat. Poster presented at the American Society of Human Genetics conference, Salt Lake City, 2005.
Klarner B, Klünemann HH, Lurding R, Aslanidis C, Rupprecht R. Neuropsychological profile of adult patients with Niemann-Pick C1 (NPC1) mutations. J Inherit Metab Dis. 2007;30:60–7.
Heitz C, Epelbaum S, Nadjar Y. Cognitive impairment profile in adult patients with Niemann pick type C disease. Orphanet J Rare Dis. 2017;12(1):166.
Johnen A, Pawlowski M, Duning T. Distinguishing neurocognitive deficits in adult patients with NP-C from early onset Alzheimer's dementia. Orphanet J Rare Dis. 2018;13(1):91.
Bergeron D, Poulin S, Laforce R Jr. Cognition and anatomy of adult Niemann-Pick disease type C: insights for the Alzheimer field. Cogn Neuropsychol. 2017;35:1–14.
Balázs N, Milanovich D, Hornyák C, Bereczki D, Kovács T. Late-onset Niemann-Pick disease type C overlapping with frontotemporal dementia syndromes: a case report. J Neural Transm (Vienna). 2019;126(11):1501–4.
Hendriksz CJ, Anheim M, Bauer P, Bonnot O, Chakrapani A, Corvol JC, et al. The hidden Niemann-Pick type C patient: clinical niches for a rare inherited metabolic disease. Curr Med Res Opin. 2017;33(5):877–90.
Micheli F, Perandones C, Giugni J, et al. Is progressive supranuclear palsy part of the phenotypic spectrum of Niemann-Pick disease type C? Neurology. 2013;80:P04.158.
Zech M, Nübling G, Castrop F, Jochim A, Schulte EC et al. Niemann-Pick C disease gene mutations and age-related neurodegenerative disorders. PLoS ONE 2013;8(12): e82879.
Boenzi S, Dardis A, Russo P, Bellofatto M, Imbriglio T, Fico T, et al. Screening for Niemann-Pick type C disease in neurodegenerative diseases. J Clin Neurosci. 2019;68:266–7.
Kluenemann HH, Nutt JG, Davis MY, Bird TD. Parkinsonism syndrome in heterozygotes for Niemann-Pick C1. J Neurol Sci. 2013;335(1–2):219–20.
Schneider SA, Tahirovic S, Hardy J, Strupp M, & Bremova-Ertl T. Do heterozygous mutations of Niemann–Pick type C predispose to late-onset neurodegeneration: a review of the literature. J Neurol 2019.
Evans WRH, Hendriksz CJ. Niemann–Pick type C disease – the tip of the iceberg? A review of neuropsychiatric presentation, diagnosis and treatment. BJPsych Bull. 2017;41(2):109–14.
Mengel E, et al. Differences in Niemann-Pick disease type C symptomatology observed in patients of different ages. Genét Mol Metab. 2017;120:80–189.
Walterfang M, Macfarlane MD, Looi JC, et al. Pontine-to-midbrain ratio indexes ocular-motor function and illness stage in adult Niemann-Pick disease type C. Eur J Neurol. 2012;19(3):462–7.
Walterfang M, Fahey M, Abel L, Fietz M, Wood A, Bowman E, et al. Size and shape of the corpus callosum in adult Niemann-Pick type C reflects state and trait illness variables. AJNR Am J Neuroradiol. 2011;32(7):1340–6.
Gburek-Augustat J, Groeschel S, Kern J, Beck-Woedl S, Just J, Harzer K, et al. Comparative analysis of cerebral magnetic resonance imaging changes in nontreated infantile, juvenile and adult patients with Niemann-Pick disease type C. Neuropediatrics. 2019.
Masingue M, Adanyeguh I, Nadjar Y, Sedel F, Galanaud D, Mochel F. Evolution of structural neuroimaging biomarkers in a series of adult patients with Niemann-Pick type C under treatment. Orphanet J Rare Dis. 2017;12(1):22.
Scheel M, Abegg M, Lanyon LJ, Mattman A, Barton JJ. Eye movement and diffusion tensor imaging analysis of treatment effects in a Niemann-Pick Type C patient. Mol Genet Metab. 2010;99(3):291–5.
Walterfang M, Fahey M, Desmond P, Wood A, Seal ML, Steward C, et al. White and gray matter alterations in adults with Niemann-Pick disease type C: a cross-sectional study. Neurology. 2010;75(1):49–56.
Kumar A, Chugani HT. Niemann-Pick disease type C: unique 2-deoxy-2[18F] fluoro-D-glucose PET abnormality. Pediatr Neurol. 2011;44(1):57–60.
Tedeschi G, Bonavita S, Barton NW, Bertolino A, Frank JA, Patronas NJ, et al. Proton magnetic resonance spectroscopic imaging in the clinical evaluation of patients with Niemann-Pick type C disease. J Neurol Neurosurg Psychiatry. 1998;65(1):72–9.
Galanaud D, Tourbah A, Lehéricy S, Leveque N, Heron B, Billette de Villemeur T, et al. 24 month-treatment with miglustat of three patients with Niemann-Pick disease type C: follow up using brain spectroscopy. Mol Genet Metab. 2009;96(2):55–8.
Benussi A, Cotelli MS, Padovani A, Borroni B. Recent neuroimaging, neurophysiological, and neuropathological advances for the understanding of NPC. F1000Res. 2018;7:194.
Esposito M, Dubbioso R, Tozza S, Iodice R, Aiello M, Nicolai E, et al. Evidência in vivo de deposição de amilóide cortical na forma adulta de Niemann Pick tipo C. Heliyon. 2019;5(11):e02776.
Saito Y, Suzuki K, Nanba E, Yamamoto T, Ohno K, Murayama S. NiemannPick type C disease: accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity. Ann Neurol. 2002;52:351–5.
Bonnot O, Klünemann HH, Velten C, Torres Martin JV, Walterfang M. Systematic review of psychiatric signs in Niemann-Pick disease type C. World J Biol Psychiatry. 2019;20(4):320–32.
Bonnot O, Klunemann HH, Sedel F, Tordjman S, Cohen D, Walterfang M. Diagnostic and treatment implications of psychosis secondary to treatable metabolic disorders in adults: a systematic review. Orphanet J Rare Dis. 2014;9:65.
Walterfang M, et al. The neuropsychiatry of Niemann-Pick type C disease in adulthood. J Neuropsychiatry Clin Neurosci. 2007;218:2.
Sevin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, et al. The adult form of Niemann-Pick disease type C. Brain. 2007;130:120–33.
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106(3):330–44.
Bauer P, Balding DJ, Klünemann HH, Linden DE, Ory DS, Pineda M, et al. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study. Hum Mol Genet. 2013;22:4349–56.
Maubert A, Hanon C, Sedel F. Psychiatric disorders in adult form of NiemannPick disease type C. Encephale. 2016;42:208–13.
Imrie J, Vijayaraghaven S, Whitehouse C, Harris S, Heptinstall L, Church H, et al. Niemann-Pick disease type C in adults. J Inherit Metab Dis. 2002;25(6):491–500.
Klünemann HH, Santosh PJ, Sedel F. Treatable metabolic psychoses that go undetected: what Niemann-Pick type C can teach us. Int J Psychiatry Clin Pract. 2012;16:162–9.
Friston K, Frith C. Schizophrenia: a disconnection syndrome? Clin Neurosci. 1995;3:89–97.
Rego T, Farrand S, Goh AMY, Eratne D, Kelso W, Mangelsdorf S, et al. Psychiatric and cognitive symptoms associated with Niemann-Pick type C disease: neurobiology and management. CNS Drugs. 2019;33(2):125–42.
Davidson CD, Walkley SU. Niemann-Pick type C disease – pathophysiology and future perspectives for treatment. US Neurol Touch Brief. 2010;6:88–94.
Eratne D, Loi SM, Li QX, Varghese S, McGlade A, Collins S, et al. Cerebrospinal fluid neurofilament light chain is elevated in Niemann-Pick type C compared to psychiatric disorders and healthy controls and may be a marker of treatment response. Aust N Z J Psychiatry. 2019;4867419893431.
Vankova J, Stepanova I, Jech R, Elleder M, Ling L, Mignot E, et al. Sleep disturbances and hypocretin deficiency in Niemann-Pick disease type C. Sleep. 2003;26(4):427–30.
Nevsimalova S, Malinova V. Cataplexy and sleep disorders in Niemann-Pick type C disease. Curr Neurol Neurosci Rep. 2015;15(1):522.
Kawazoe T, Yamamoto T, Narita A, Ohno K, Adachi K, Nanba E, et al. Phenotypic variability of Niemann-Pick disease type C including a case with clinically pure schizophrenia: a case report. BMC Neurol. 2018;18:117.
Harzer K, Beck-Wödl S, Bauer P. Niemann-pick disease type C: new aspects in a long published family - partial manifestations in heterozygotes. JIMD Rep. 2014;12:25–9.
Reading P. Cataplexy. Pract Neurol. 2019 Feb;19(1):21–7.
Patterson M. Niemann-pick disease type C. 2000 Jan 26 [Updated 2019 Aug 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019.
Pedroso JL, Fusão EF, Ladeia-Frota C, Arita JH, Barsottini OG, Masruha MR, et al. Teaching video neuroimages: gelastic cataplexy as the first neurologic manifestation of Niemann-Pick disease type C. Neurology. 2012 Nov 27;79(22):e189.
Canto CB, Onuki Y, Bruinsma B, van der Werf YD, De Zeeuw CI. The sleeping cerebellum. Trends Neurosci. 2017;40(5):309–23.
Xu S, Zhou S, Xia D, Xia J, Chen G, Duan S, et al. Defects of synaptic vesicle turnover at excitatory and inhibitory synapses in Niemann-Pick C1-deficient neurons. Neuroscience. 2010;167(3):608–20.
Sáez PJ, Orellana JA, Vega-Riveros N, Figueroa VA, Hernández DE, Castro JF, et al. Disruption in connexin-based communication is associated with intracellular Ca2+ signal alterations in astrocytes from Niemann-pick type C mice. PLoS One. 2013;8(8):e71361.
Stampfer M, Theiss S, Amraoui Y, Jiang X, Keller S, Ory DS, et al. Niemann-Pick disease Type C clinical database: cognitive and coordination deficits are early disease indicators. Orphanet J Rare Dis. 2013;8(1):35.
Munoz T, Raiman J. Epilepsy and Niemann Pick C disease. J Pediatric Epilepsy. 2015;03(04):229–34.
Skorpen J, Helland IB, Tennøe B. Use of miglustat in a child with late-infantile-onset Niemann-Pick disease type C and frequent seizures: a case report. J Med Case Rep. 2012;6:383.
Higgins JJ, Patterson MC, Dambrosia JM, Pikus AT, Pentchev PG, Sato S, et al. A clinical staging classification for type C Niemann-Pick disease. Neurology. 1992;42(12):2286–90.
Santos ML, Raskin S, Telles DS, Lohr A Jr, Liberalesso PB, Vieira SC, et al. Treatment of a child diagnosed with NiemannPick disease type C with miglustat: a case report in Brazil. J Inherit Metab Dis. 2008;31(Suppl 2):S357–61.
Karimzadeh P, Tonekaboni SH, Ashrafi MR, Shafeghati Y, Rezayi A, Salehpour S, et al. Effects of miglustat on stabilization of neurological disorder in niemann-pick disease type C: Irania. J Child Neurol. 2013;28(12):1599–606.
Heron B, Valayannopoulos V, Baruteau J, Chabrol B, Ogier H, Latour P, et al. Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C. Orphanet J Rare Dis. 2012;7:36.
Wijburg FA, Sedel F, Pineda M, Hendriksz CJ, Fahey M, Walterfang M, et al. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology. 2012;78(20):1560–7.
Mengel E, Klünemann HH, Lourenço CM, Hendriksz CJ, Sedel F, Walterfang M, et al. Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J Rare Dis. 2013;8:166.
Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009;98:152–65.
Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J Rare Dis. 2018;13:140.
Bonney DK, O’Meara A, Shabani A, Imrie J, Bigger BW, Jones S, et al. Successful allogeneic bone marrow transplant for Niemann-Pick disease type C2 is likely to be associated with a severe ‘graft versus substrate’ effect. J Inherit Metab Dis. 2010;3:171–3.
Senirli RT, Kuşçu O, Akyol U, Topçu M, Yiğit Ö, Aksoy S, et al. Otorhinolaryngological, audiovestibular and swallowing manifestations of patients with Niemann–Pick disease type C. Int J Pediatr Otorhinolaryngol. 2016;80:1–4.
Yanjanin NM, Velez JI, Gropman A, King K, Bianconi SE, Conley SK, et al. Linear clinical progression, independent of age of onset, in Niemann-Pick disease, type C. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:132–40.
King KA, Gordon-Salant S, Yanjanin N, Zalewski C, Houser A, Porter FD, et al. Auditory phenotype of Niemann-Pick disease, type C1. Ear Hear. 2014;35(1):110–7.
Guo W, He A, Boer M. Elevated plasma chitotriosidase activity in various Lysosomal storage disorders. J Inherit Metab Dis. 1995;18(6):717–22.
Ward S, O’Donnell P, Fernandez S, et al. 2-Hydroxypropyl-β-Cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr Res. 2010;68(1):52–6.
Belmatoug N, Burlina A, Giraldo P, Hendriksz CJ, Kuter DJ, Mengel E, et al. Gastrointestinal disturbances and their management in miglustat-treated patients. J Inherit Metab Dis. 2011;34(5):991–1001.
Copetti S D, Santos G N. Feeding for patients with neurodegenerative disoders as a focus on Niemann type C Pick disease (Npd). Nov Tech Nutri Food Sci. 2(2). NTNF.000535. 2018.
Coisne C, Tilloy S, Monflier E, Wils D, Fenart L, Gosselet F. Cyclodextrins as emerging therapeutic tools in the treatment of cholesterol-associated vascular and neurodegenerative diseases. Molecules. 2016;21:E1748.
Och U, Fischer T, Marquardt T. Dietary carbohydrate modification in Niemann-Pick type C. Case series of dietary treatment during miglustat (Zavesca®) therapy. Ernährungs Umschau. 2019;66(03):36–44.
Papandreou A, Gissen P. Diagnostic workup and management of patients with suspected Niemann-Pick type C disease. Ther Adv Neurol Disord. 2016;9(3):216–29.
Telles-Correia D, Barbosa-Rocha N, Gama-Marques J, Moreira AL, Alves-Moreira C, Saraiva S, et al. Validation of the Portuguese version of the Psychotic Symptom Rating Scales (PSYRATS). Actas Esp Psiquiatr. 2017;45(2):56–61.
Vessoni AL. Adaptação e estudo de confiabilidade da escala de avaliação das síndromes positiva e negativa para a esquizofrenia no Brasil [dissertação]. São Paulo: Escola Paulista de Medicina; 1993.
Bressan RA, Chaves AC, Shirakawa I, de Mari J. Validity study of the Brazilian version of the Calgary Depression Scale for Schizophrenia. Schizophr Res. 1998;32(1):41–9.
Arciniegas DB. Psychosis. Continuum (Minneap Minn). 2015;21(3 Behavioral Neurology and Neuropsychiatry)2015;21(3):715-736.
Bertolazi AN, Fagondes SC, Hoff LS, Pedro VD, Menna Barreto SS, Johns MW. Portuguese-language version of the Epworth sleepiness scale: validation for use in Brazil. J Bras Pneumol. 2009;35(9):877–83.
Bertolazi AN, Fagondes SC, Hoff LS, Dartora EG, Miozzo IC, de Barba ME, et al. Validation of the Brazilian Portuguese version of the Pittsburgh Sleep Quality Index. Sleep Med. 2011;12(1):70–5.
Masuko AH, Carvalho LB, Machado MA, Morais JF, Prado LB, Prado GF. Translation and validation into the Brazilian Portuguese of the restless legs syndrome rating scale of the International Restless Legs Syndrome Study Group. Arq Neuropsiquiatr. 2008;66(4):832–6.
da Silva TI, Alonso NB, Azevedo AM, Westphal-Guitti AC, Migliorini RCVP, Marques CM, et al. Tradução e adaptação cultural da Seizure Severity Questionnaire: resultados preliminares. J Epilepsy Clin Neurophysiol. 2006;12(1):41–7.
Bowman EA, Walterfang M, Abel L, Desmond P, Fahey M, Velakoulis D. Longitudinal changes in cerebellar and subcortical volumes in adult-onset Niemann-Pick disease type C patients treated with miglustat. J Neurol. 2015;262:2106–14.
Szakszon K, Szegedi I, Magyar Á, Oláh É, Andrejkovics M, Balla P, et al. Complete recovery from psychosis upon miglustat treatment in a juvenile Niemann-Pick C patient. Eur J Paediatr Neurol. 2014;18:75–8.
Santos MLF, Raskin S, Telles DS, Löhr Junior A, Liberalesso PBN, Vieira SC, et al. Treatment of a child diagnosed with Niemann-Pick disease type C with miglustat: a case report in Brazil. J Inherit Metab Dis. 2008;31:357–61.
Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007;6:765–72.
Liu B. Therapeutic potential of cyclodextrins in the treatment of Niemann-Pick type C disease. Clin Lipidol. 2012;7:289–301.
Berry-Kravis E, Chin J, Hofmann A, Winston A, Stoner R, LaGorio L, et al. Long-term treatment of Niemann-Pick type C1 disease with intrathecal 2-hydroxypropyl-β-cyclodextrin. Pediatr Neurol. 2018;80:24–34.
Megías-Vericat JE, García-Robles A, Company-Albir MJ, Fernández-Megía MJ, Pérez-Miralles FC, López-Briz E, et al. Early experience with compassionate use of 2 hydroxypropyl-beta-cyclodextrin for Niemann-Pick type C disease: review of initial published cases. Neurol Sci. 2017;38:727–43.
Kirkegaard T, Gray J, Priestman DA, et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci Transl Med. 2016;8(355):355ra118.
Subramanian K, Hutt DM, Scott SM, Gupta V, Mao S, Balch WE. Correction of Niemann-Pick type C1 trafficking and activity with the histone deacetylase inhibitor valproic acid. J Biol Chem. 2020:jbc.RA119.010524.
Bremova T, Malinová V, Amraoui Y, Mengel E, Reinke J, Kolníková M, et al. Acetyl-dl-leucine in Niemann-Pick type C: a case series. Neurology. 2015;85(16):1368–75.
Schultz ML, Krus KL, Lieberman AP. Lysosome and endoplasmic reticulum quality control pathways in Niemann-Pick type C disease. Brain Res. 1649;2016:181–8.
Funding
This study was not supported by any specific grant from funding agencies in the public, commercial, or non-profit sectors.
Author information
Authors and Affiliations
Contributions
Conception and design of the work: DMR and PBN. SSOC: literature search; acquisition, analysis, or interpretation of data for the work: DMR, MASN, and PBN. Drafting the work: DMR, JLP, MASN, and PBN. All authors were involved in critical revision of the manuscript for important intellectual content.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Statement
Patients signed an informed consent and allowed publication of this data.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rangel, D.M., Melo, M.C.A., Pedroso, J.L. et al. Beyond the Typical Syndrome: Understanding Non-motor Features in Niemann-Pick Type C Disease. Cerebellum 19, 722–738 (2020). https://doi.org/10.1007/s12311-020-01156-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-020-01156-0