
Abstract
Background and Objective
Daridorexant is a dual orexin receptor antagonist in clinical development for insomnia. As daridorexant is cleared mainly via cytochrome P450 (CYP) 3A4, the effect of hepatic impairment on the pharmacokinetics (PK), metabolism, and tolerability of daridorexant was evaluated. Sleep disorders are common in patients with liver cirrhosis and, therefore, sleep-promoting drugs with a better tolerability than currently available would be preferable, a premise that dual orexin receptor antagonists may fulfill.
Methods
This was a single-dose, open-label, phase I study. Subjects with mild (Child–Pugh A, N = 8) or moderate (Child–Pugh B, N = 8) liver cirrhosis and matched healthy control subjects (N = 8) received 25 mg of daridorexant orally. Blood samples were collected for 72 h post-dose for PK assessments of daridorexant and three major metabolites.
Results
Compared with healthy subjects, patients showed a decrease in total daridorexant area under the plasma concentration–time curve from zero to infinity (AUC0-inf) and maximum plasma concentration with a geometric mean ratio (GMR, 90% confidence interval [CI]) of 0.51 (0.28–0.92) and 0.50 (0.35–0.72) in Child–Pugh A and 0.74 (0.39–1.41) and 0.42 (0.29–0.60) in Child–Pugh B patients, respectively. Furthermore, the median time to reach maximum plasma concentration was slightly delayed (1.0 h [90% CI 0.0–2.0] in Child–Pugh A patients and 0.5 h [90% CI 0.0–1.5] in Child–Pugh B patients), while for Child–Pugh B patients, a doubling in half-life was observed (GMR [90% CI]: 2.09 [1.32–3.30]). Considering the high plasma protein binding (> 99%) and a 1.9-fold to 2.3-fold increase in the unbound fraction in patients, the PK of unbound daridorexant was also assessed. Compared with healthy subjects, Child–Pugh B patients had a higher AUC0-inf (GMR [90% CI] 1.60 [0.93–2.73]), a lower apparent plasma clearance (GMR [90% CI] 0.63 [0.37–1.07]), and the same doubling in the half-life observed for total daridorexant, whereas maximum plasma concentration and apparent volume of distribution were not different. Unbound daridorexant PK in Child–Pugh A patients did not differ from healthy subjects. In addition, the metabolic ratios (parent to metabolite), i.e., a marker of CYP 3A4 activity, of the two most abundant daridorexant metabolites were higher in patients with liver cirrhosis compared with healthy subjects. All treatment-emergent adverse events were transient and of mild or moderate intensity and no other treatment-related effects were apparent.
Conclusions
No safety issue of concern was detected following administration of 25 mg of daridorexant in the study population. Moderate liver cirrhosis causes impaired hepatic clearance of unbound daridorexant, which prolongs the half-life. A 25-mg dose of daridorexant should, therefore, not be exceeded in Child–Pugh B patients. A dose adjustment is not required in Child–Pugh A patients, while avoidance of daridorexant in patients with Child–Pugh C cirrhosis is recommended.
Clinical Trial Registration
ClinicalTrials.gov ID: NCT03713242.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Daridorexant, a potent and selective dual orexin receptor antagonist being developed to treat insomnia, has been shown to have a desired effect on sleep onset and sleep maintenance in phase II and III studies. |
Previous phase I studies have demonstrated that daridorexant is mainly metabolized by cytochrome P450 3A4. |
By evaluating the tolerability and pharmacokinetics of total and unbound daridorexant (as well as of three major metabolites) following oral administration of 25 mg of daridorexant in healthy subjects and patients with mild and moderate hepatic impairment (Child–Pugh A and B), this study enabled dosing recommendations for patients with liver cirrhosis. |
The study shows the importance of determining the pharmacokinetics of the unbound fraction of highly protein-bound drugs in patients with liver cirrhosis. |
1 Introduction
Orexins A and B were detected in 1998 and identified as peptides binding to two G-protein-coupled receptors [1]. Orexin A and B are peptides synthesized by orexin neurons, which originate in the lateral hypothalamic area and posterior hypothalamus and project into the entire central nervous system (CNS) excluding the cerebellum [2]. Importantly, they activate the monoaminergic and cholinergic nuclei in the brainstem and hypothalamus and thereby regulate sleep and wakefulness. Orexins act on orexin receptors, G-protein-coupled transmembraneous proteins that increase the intracellular calcium level when activated by binding orexin A or B. Orexin receptors are distributed in the entire CNS, but are found with a high density in the paraventricular nucleus of the thalamus, the locus coeruleus, and the dorsal raphe nucleus of the brainstem [2,3,4]. Two orexin receptors have been described, orexin-1 and orexin-2 (OX1 and OX2 receptors), which are expressed at similar sites in the CNS [2, 5]. While OX1 receptors have a higher affinity for orexin A than B, OX2 receptors have a similar affinity for both orexins [6].
Considering that the main function of orexins is to promote wakefulness [2], the blocking of orexin receptors has become a target of intense interest for the development of novel sleep medications [7, 8]. Accordingly, suvorexant, the first-in-class dual orexin receptor antagonist (DORA) was approved by the US Food and Drug Administration and the Japanese Health Authorities as a hypnotic agent in 2014, while lemborexant has been approved more recently [5].
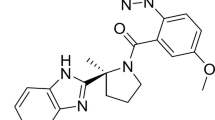
Daridorexant (ACT-541468) is a DORA for which several phase I and II trials have been published and phase III trials have been concluded, wherein daridorexant was well tolerated with positive effects on sleep parameters and without next-morning residual effects [9,10,11,12,13,14,15,16]. Daridorexant has an absolute bioavailability of 62% and a terminal half-life (t½) of approximately 8 h. Following administration of 14C-labeled daridorexant, the cumulative recovery of radioactivity from feces and urine was 85%, with fecal excretion being the major route of elimination [15]. Daridorexant is heavily metabolized in the liver with more than 70 identified metabolites [17]. In plasma, the most abundant metabolites, which do not significantly contribute to the pharmacological effect, are a mono-oxidized benzylic alcohol (ACT-776063 [M3]), the corresponding aldehyde (ACT-776537 [M1]), and ACT-1016-3307 (M10) (Fig. 1). Preclinical experiments have shown that cytochrome P450 (CYP) 3A4 accounts for approximately 90% of total turnover in vitro with only minor contributions from other enzymes [12, 17].

Adapted from Muehlan et al. [17]
Chemical structure of daridorexant and its major metabolites. M3 is a primary alcohol formed by hydroxylation of the methyl group at the benzimidazole ring. This reaction is catalyzed mainly by CYP 3A4. M3 is the origin of M1, which is formed by oxidation of the primary alcohol to the corresponding aldehyde, and of M10, which is also obtained by an oxidative rearrangement of metabolite M5.
In patients with liver cirrhosis, benzodiazepines or benzodiazepine-like drugs (z-drugs) are particularly problematic because of the risk of developing hepatic encephalopathy [18, 19]. As sleep disorders are common in this population, sleep-promoting drugs with a better tolerability would be preferable. As DORAs could fulfill this premise, the pharmacokinetics (PK), metabolism, and tolerability of daridorexant in patients with mild (Child–Pugh A) or moderate (Child–Pugh B) liver cirrhosis were investigated. Thereby, the influence of plasma protein binding on the PK of daridorexant was investigated specifically.
2 Methods
2.1 Clinical Study
This single-center, open-label, single-dose, phase I study was planned to be conducted with patients from all three Child–Pugh categories (mild [Group A], moderate [Group B], and severe [Group C]; N = 8 per group) as well as control subjects with normal hepatic function (Group D; matched to the group with the most severe hepatic impairment; N = 8) [20, 21]. Screening assessments were performed between 1 and 28 days prior to dosing.
The subjects stayed in the clinic until 24 h post-dose PK blood sampling and safety assessments had been performed, upon which they could be discharged based on their medical condition. An end of study (EOS) examination took place 72 h after dosing for subjects with hepatic impairment (48 h post-dose for healthy subjects). For safety considerations, dosing in subjects with mild (Group A), moderate (Group B), and severe (Group C) hepatic impairment was to be staggered, thus dosing of Group A started first. After an interim analysis of PK, safety, and tolerability data of at least six subjects with hepatic impairment from the previous group, dosing of the next group was started.
The study (ClinicalTrials.gov identifier: NCT03713242) was conducted at the University Hospital Basel, Basel, Switzerland, and was approved by both the National Health Authority of Switzerland (Swissmedic) and the local ethics committee (Ethikkommission Nordwest- und Zentralschweiz, Basel, Switzerland). The study adhered to the Declaration of Helsinki and was conducted according to good clinical practice. Prior to any study procedure, written informed consent was obtained from each participant after adequate explanation of the objectives, methods, and potential hazards of the study.
2.2 Subjects and Treatment
Healthy subjects (Group D) were defined as such based on physical examination, medical history, 12‐lead electrocardiogram (ECG), routine hematology, clinical chemistry, and urinalysis and were matched to subjects in the group with the most severe hepatic impairment studied regarding sex, age (± 10 years difference allowed), body weight, and height (± 15% difference allowed). The minimum creatinine clearance (according to the Cockcroft–Gault formula) for inclusion was stratified by age and was > 80 mL/min for subjects < 50 years of age, > 70 mL/min for subjects 50–60 years of age, and > 60 mL/min for subjects 61–75 years of age [22]. Subjects with clinical evidence of any disease or any surgical or medical condition with a potential to interfere with the absorption, distribution, metabolism, or excretion of the study drug except for those related to liver cirrhosis (e.g., herniotomy, cholecystectomy allowed) were not enrolled in the study. Further, subjects with active diseases of the CNS, chronic symptoms of pronounced constipation or diarrhea, symptoms of a clinically relevant illness, recent gastrointestinal bleeding, history of stroke, myocardial infarction, clinically relevant stenosis of the vessels supplying the brain, tense ascites, or pleural effusion were excluded from study participation. Abdominal sonography was carried out to grade ascites and to exclude the presence of hepatocellular carcinoma. Liver cirrhosis was diagnosed based on a liver biopsy in all subjects. Subjects with encephalopathy grade > 2 were excluded from the study. Concomitant use of medication was prohibited in healthy subjects except for hormonal contraceptives and medications for the treatment of adverse events (AEs). Patients with hepatic impairment could continue taking their regularly prescribed medications, except for medication that might influence results of the trial (e.g., CYP3A4 inhibitors or inducers). Subjects with a history (healthy subjects only) or clinical evidence of alcoholism or drug abuse were excluded from participation. Furthermore, consumption of grapefruit and/or Seville orange fruit and juice was not permitted from screening until the EOS visit. Smoking was restricted during the in-clinic stay, as was drinking of alcoholic beverages from at least 48 h prior to clinic admission until EOS.
A single oral dose of 25 mg of daridorexant was selected as it represents the mid-range dose investigated in the phase III studies (i.e., 10, 25, and 50 mg were evaluated) and was shown to be well tolerated and efficacious based on data from previous phase I and II studies, allowing for an accurate investigation of the single-dose PK, tolerability, and safety [9,10,11]. Subjects remained fasted from at least 10 h prior to study drug administration until 2 h thereafter. Fluid intake was not allowed from 1 h before until 1 h after study drug administration. Each subject received a single oral dose of 25 mg of daridorexant in the morning, which was administered with 240 mL of water. The subjects remained in bed in a sitting position from approximately 5 min before until 2 h after intake of the study drug, except for the measurement of vital signs and ECG assessments (conducted in the supine position).
2.3 Blood Sampling and Bioanalysis
PK blood samples (3 mL) were collected into potassium ethylenediamine-tetra acetic acid-containing tubes from an indwelling catheter or by direct venipuncture pre-dose and at scheduled intervals at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, and 48 h (healthy subjects) or up to 72 h post-dose (patients with liver cirrhosis). For determination of the extent of plasma protein binding of daridorexant, blood samples were taken at 1 h and 3 h post-dose, i.e., in the range covering the expected time to maximum plasma concentration (tmax) of daridorexant.
Plasma concentrations of daridorexant and metabolites (M1, M3, and M10) were measured using two validated liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) assays [14, 15]. The limit of quantification (LOQ) was 0.5 ng/mL for daridorexant and 2.0 ng/mL for the three metabolites of daridorexant with both methods covering a range up to 2000 ng/mL. The inter-batch precision for daridorexant was ≤ 6.0% and the accuracy was − 1.4% to 3.6%. The inter-batch precision for the three metabolites together was ≤ 4.6% and the accuracy was − 3.3 to 3.6%.
The unbound fraction (Cu/C) of daridorexant in plasma was determined using an equilibrium dialysis followed by analysis with a validated LC-MS/MS method slightly adapted from the one described above. Triplicate 200-µL aliquots of each plasma sample were subjected to rapid equilibrium dialysis (Thermo Fisher Scientific, Waltham, MA, USA) against 350 µL of fortified phosphate-buffered saline (PBS) on an orbital shaker at 450 rpm incubated at 37 °C for 5 h. After dialysis, a 50-μL aliquot of the donor compartment was diluted with 50 μL of PBS, while a 50-μL aliquot of the receiver compartment was diluted with 50 μL of blank plasma, to generate samples with the same analytical matrix. The LC-MS/MS method was linear in the concentration range of 0.1–400 ng/mL with an LOQ of 0.1 ng/mL. The inter-batch precision was ≤ 5.0% with an inter-batch accuracy of − 5.0% to 5.0%.
2.4 Pharmacokinetic Evaluations and Statistics
PK parameters of daridorexant (total and unbound) and of the three major daridorexant metabolites were obtained by a non-compartmental analysis using Phoenix WinNonlin (Version 8.0; Certara, Princeton, NJ, USA). Measured individual plasma concentrations were used to directly obtain maximum plasma concentration (Cmax) and tmax. Unbound daridorexant concentrations were derived from the measured individual total daridorexant concentrations and the respective mean of Cu/C measured at 1 h and 3 h post-dose for each subject. Area under the plasma concentration–time curve (AUC) from time zero to infinity (AUC0-inf) was calculated by combining AUC from zero to time t of the last concentration above the LOQ according to the linear trapezoidal rule and AUCextra obtained by Ct/λz (AUCextra). The Ct was the last plasma concentration above the LOQ and λz was the terminal elimination rate constant determined by log-linear regression analysis of the plasma concentrations of the terminal elimination phase. The t½ was calculated as ln(2)/λz. Concentrations below LOQ were entered as zero and included in the calculation of means. For plasma protein binding, Cu/C was expressed as a percentage. Metabolic ratios (MRs) were calculated as the ratio of AUC0–48h of total daridorexant and AUC0–48 of the respective metabolite [23]. The PK parameters Cmax, AUC, apparent plasma clearance (CL/F, with F indicating bioavailability), apparent volume of distribution (Vz/F), and t½ as well as Cu/C were summarized using geometric means and their two-sided 95% confidence interval (CI). Median and range values were used for tmax. Differences in Cmax, AUC, CL/F, Vz/F, t½, and Cu/C between healthy subjects and hepatic impairment groups were explored using their geometric mean ratios and 90% CI with healthy subject results as the reference. Differences between treatments for tmax were explored using the non-parametric Wilcoxon signed rank test and Hodges–Lehmann estimates of the median of differences and their 90% CIs. Correlations between the free fraction of daridorexant and the Vz/F or the α1-acid glycoprotein (α1-AGP) plasma level were investigated with Spearman’s correlation analysis. The sample size of eight subjects per group was based on empirical considerations.
2.5 Safety Assessments
The safety and tolerability of the study drug were evaluated throughout the study on the basis of reported AEs, results of physical examination, assessment of body weight, vital signs (blood pressure and pulse rate), 12-lead ECGs, and clinical laboratory tests (hematology, clinical chemistry [including α1-AGP], coagulation, and urinalysis) and were analyzed descriptively. The Karolinska Sleepiness Scale (KSS) was used to assess the alertness of the subjects 24 h post-dose [24].
3 Results
3.1 Demographics
Overall, 24 subjects (11 male, 13 female) were included in three groups and completed the study as per protocol between January 2019 and February 2020. Based on the results of the interim PK data analysis following investigation of the subjects with moderate hepatic impairment, i.e., doubling in t½ compared with healthy subjects (see Table 2), no subjects with severe hepatic impairment (Group C) were dosed. Chronic alcohol abuse and chronic hepatitis C virus infection were identified as the primary etiology for liver cirrhosis in most patients. The three groups were well matched for age, body weight, height, and body mass index (Table 1). Six subjects (6/8 [75.0%]) in Group A and eight subjects (8/8 [100%]) in Group B reported stable use of concomitant medications for the treatment of hepatic and other metabolic disorders. All concomitant medications were compliant with the study requirements.
3.2 Pharmacokinetics
The plasma concentration–time profile of total daridorexant in healthy subjects (Group D) was characterized by a Cmax of 846 ng/mL (geometric mean) attained at a median tmax of 1.0 h (Fig. 2 and Table 2). The AUC0–inf was 6707 ng·h/mL, and the t½ was 11.0 h. In addition, a CL/F of 3.73 L/h and a Vz/F of 59.0 L was observed (Table 2). Compared with healthy subjects, a decrease in Cmax [geometric mean (90% CI)] of daridorexant by 50% (0.35–0.72) in Group A and by 58% (0.29–0.60) in Group B was observed (Table 2). The median tmax was reached later in patients with hepatic impairment than in healthy subjects, i.e., median difference (90% CI) was 1.0 h (0.0–2.0) in Group A and 0.5 h (0.0–1.5) in Group B. The AUC0-inf was decreased by 49% (0.28–0.92) and 26% (0.39–1.41) compared with Group D in Groups A and B, respectively. The elimination phase was characterized by an unchanged t½ in Group A [geometric mean ratio 0.98 (0.62–1.55)] and by a 2.1-fold (1.32–3.30) increase in Group B. In Groups A and B, the CL/F as well as the Vz/F of daridorexant were increased compared with Group D; CL/F by 1.97-fold (1.09–3.58) and 1.35-fold (0.71–2.58) and Vz/F by 1.93-fold (1.34–2.78) and 2.04-fold (1.37–3.03), respectively.

Plasma concentration–time profile of total daridorexant. The concentration data were obtained after an oral dose of 25 mg and are presented as mean ± standard deviation. The calculated pharmacokinetic parameters are given in Table 2
The Cu/C of daridorexant in healthy subjects at 1 h and 3 h post-dose was 0.18% and 0.15%, respectively (Table 2). An increase in Cu/C by 1.9-fold in Group A and by 2.3-fold in Group B was measured (average of Cu/C measured 1 h and 3 h post-dose). Spearman’s correlation was run to determine the relationship between Cu/C and total daridorexant Vz/F as well as α1-AGP plasma levels, which revealed a positive correlation with Vz/F (r = 0.844, p < 0.0001; Fig. 3a) and a negative correlation with α1-AGP (r = −0.888, p < 0.0001; Fig. 3b).

Correlation of the free fraction of daridorexant with the apparent volume of distribution (Vz/F) of total daridorexant (a) and with the plasma α1-acid glycoprotein level (b). The correlation coefficient r is 0.844 for a and −0.888 for b
The plasma concentration–time curve (Fig. 4) and the PK parameters of unbound daridorexant in subjects with mild hepatic impairment (Group A) and healthy subjects were similar with only a delay in tmax apparent (Table 2). In subjects with moderate hepatic impairment (Group B), Cmax remained unchanged compared to healthy subjects, while an increase in AUC0–inf by 1.6-fold (0.93–2.73), a decrease in unbound CL/F by 37% (0.36–1.07), and a 2.1-fold (1.32–3.30) prolongation in t½ could be observed.

Plasma concentration–time profile of unbound daridorexant. The concentration data were obtained after an oral dose of 25 mg and are presented as mean ± standard deviation. The calculated pharmacokinetic parameters are given in Table 2
Similar to healthy subjects, M3 was also the most predominant plasma metabolite in patients with hepatic impairment, followed by M1 and M10 ( Table 1 of the Electronic Supplementary Material [ESM]). As shown in Fig. 5 and Figs. 1, 2 of the ESM, the concentration–time profiles of all three metabolites generally matched the profile of the parent as Cmax and AUC tended to be lower in patients with liver cirrhosis compared with healthy subjects. In addition, t½ was prolonged in patients with moderately impaired liver function compared with subjects with mildly impaired or normal liver function. The MRs for the most abundant metabolites M1 and M3 were higher in patients with liver cirrhosis compared with healthy subjects (Table 1 of the ESM).

Plasma concentration–time profile of the daridorexant metabolite M3. M3 is a primary alcohol formed by hydroxylation of the methyl group at the benzimidazole ring system. CYP 3A4 is the main CYP involved in this reaction. Data are presented as the mean ± standard deviation
3.3 Safety and Tolerability
The only AEs reported during the study were somnolence (once in Group A [1/8, i.e., 12.5%] and twice in Group B [2/8, i.e., 25.0%]; all of moderate intensity), nausea (once in Group A [1/8, i.e., 12.5%]; mild in intensity), and an asymptomatic slightly prolonged QTcB interval without clinical relevance (once in Group B [1/8, i.e., 12.5%]; mild in intensity, 72 h [at EOS] after administration of study treatment). No AEs were reported in healthy subjects. All AEs were considered by the investigator as drug related and resolved without sequelae and without need for treatment. No serious AEs, no AEs leading to discontinuation, or clinically relevant treatment-related changes in clinical laboratory, vital signs, body weight, and ECG variables were observed. Furthermore, no change in the KSS score was evident 24 h post-dose when compared to measurements at baseline in any of the three treatment groups.
4 Discussion
The study’s objectives were to assess the PK and metabolism and to describe the tolerability of daridorexant in subjects with liver cirrhosis in comparison with healthy subjects. Daridorexant is a Biopharmaceutical Classification System class IIb drug with a solubility of 256 mg/L at pH 5.0 and 37 °C and a high permeability (22.6 × 10−6 cm/s) [3]. The drug is rapidly absorbed (tmax of 1 h in healthy subjects in this study), has a bioavailability of 62% [15], is highly protein bound (> 99%) [3, 25], has a volume of distribution at steady state of 31 L in healthy subjects [15], and is eliminated with a t½ of approximately 8 h [5]. PK results in healthy subjects in this study are in line with previous studies in which 25 mg daridorexant was administered, especially with the data shown by Muehlan et al. [16], wherein a similarly aged population was studied. In that study, similar to the current findings, a t½ of 10 h was observed, whereas in studies in healthy young subjects the t½ tended to be shorter.
The drug is extensively metabolized and only traces of the parent drug are eliminated by the kidney or in feces [15]. Three major metabolites have been detected in plasma, M3, M1, and M10 (Fig. 1). M3 is a primary alcohol formed by hydroxylation of the methyl group at the benzimidazole ring [17]. This reaction is mainly catalyzed by CYP3A4 [12] and may, therefore, be used as a marker of CYP3A4 activity. M3 is the origin of the aldehyde M1, which is generated by oxidation of the benzyl alcohol. M10 results from hydroxylation of the methyl group at the benzimidazole ring and by an oxidative rearrangement reaction [17]. These three metabolites have also been observed in the current study, both in healthy subjects and in patients with liver cirrhosis.
Liver cirrhosis can affect many aspects of a drug’s PK; it can delay the rate of absorption, increase bioavailability owing to portosystemic shunts (PSS), increase Vz/F owing to reduced protein binding, and impair drug clearance owing to reduced hepatic blood flow and decreased hepatic drug metabolism [26]. Delayed absorption of orally administered drugs, as observed in the current study with daridorexant, is a well-known phenomenon in patients with liver cirrhosis [27,28,29]. Typically, the amount of drug absorbed remains unchanged, but gastric emptying and the intestinal passage may be delayed [30]. As prokinetic drugs can reverse this effect, delayed absorption has a functional and not an organic cause [31], which may be due to impaired secretion and/or function of prokinetic hormones [30, 32].
Owing to the presence of PSS, bioavailability could increase from 62% in healthy subjects to 100% in patients with liver cirrhosis, which should cause an increase in Cmax and AUC [33]. Based on a previous study, it can be assumed that the shunt index (the percentage of portal blood circumventing the liver) is approximately 15% in Child–Pugh A patients and 30% in Child–Pugh B patients [33]. As shown in Table 2, for total plasma daridorexant, the opposite of what was expected was observed, namely a decrease in Cmax and AUC in both groups of patients with liver cirrhosis compared with healthy controls. Reduced exposure to the total (protein-bound and unbound) drug has been reported for other highly protein-bound drugs in patients with liver cirrhosis and is explained below [34]. The lack of increase in Cmax of total and unbound daridorexant suggests that PSS had little influence on the PK of daridorexant. This can be explained by the already high bioavailability of daridorexant in healthy controls, by the limited shunt index in patients with Child–Pugh A and B cirrhosis, and by assuming that, in contrast to systemic metabolism, pre-systemic metabolism of daridorexant is mainly intestinal and not hepatic. The drug is metabolized by CYP3A4 by approximately 90% [12], which has a high intestinal expression [35]. In contrast to hepatic pre-systemic metabolism, PSS should not affect the intestinal pre-systemic metabolism.
The Vz/F of total daridorexant was increased two-fold in both groups of patients with liver cirrhosis compared with healthy subjects. Although bioavailability of daridorexant could be increased in patients with liver cirrhosis, this potential increase is not a sufficient explanation for this finding. A more likely explanation is the reduced plasma protein binding of daridorexant observed in patients with liver cirrhosis. Daridorexant showed a plasma protein binding of > 99%, which is in line with previously published data in humans and experimental animals [3, 25]. Importantly, in both groups of patients with liver cirrhosis, the unbound fraction was approximately doubled compared with healthy controls. The positive linear correlation of the unbound fraction of daridorexant with Vz/F of total daridorexant supports the assumption that the observed increase in Vz/F in patients is due to reduced plasma protein binding. Furthermore, the negative correlation of the plasma α1-AGP level with the free fraction of daridorexant suggests that daridorexant binds at least partially to α1-AGP. Postulated mechanisms contributing to the decreased binding of lipophilic drugs to plasma proteins in patients with liver cirrhosis include a decrease in the plasma concentration of binding proteins (mainly albumin and α1-AGP), changes in the binding characteristics of plasma proteins, and competition with endogenous compounds (e.g., bile acids, bilirubin), which can displace drugs from binding sites [36, 37]. As shown in Table 1, patients with liver cirrhosis had lower albumin and α1-AGP plasma levels depending on the severity of hepatic impairment. In addition, it is well known that patients with liver cirrhosis have elevated plasma bile acid levels compared with healthy subjects owing to PSS [33]. Decreases in plasma levels of drug-binding proteins and/or drug displacement from binding sites are associated with reduced total plasma drug concentration and the increased free fraction of protein-bound drugs, whereas the free drug concentrations may remain unaltered [38,39,40]. The decreased plasma protein binding capacity observed in subjects with liver cirrhosis, therefore, plays an important role in the apparent decrease in total daridorexant exposure.
However, as it is the unbound rather than protein-bound drug in plasma that achieves equilibrium with the unbound drug at site(s) of drug action and elimination [41], the PK of unbound daridorexant has to be considered. Indeed, the decrease in hepatic metabolic capacity in subjects with liver cirrhosis only becomes apparent if unbound daridorexant plasma PK parameters are explored. In contrast to the total drug, for unbound daridorexant no significant differences in Cmax and Vz/F between patients with liver cirrhosis and control subjects were detected. In addition, patients with Child–Pugh B liver cirrhosis showed a significant increase in AUC0–inf with a corresponding decrease in CL/F compared with healthy controls. As Vz/F was not different between patients with Child–Pugh B liver cirrhosis and control subjects, the prolonged t½ of daridorexant in these patients is explained by the reduced CL/F of daridorexant. The same explanation was given for the prolonged half-life of erythromycin [39] and diazepam [42] in patients with liver cirrhosis.
Considering the bioavailability of 62% in healthy subjects, daridorexant is a low clearance drug whose hepatic clearance is mainly controlled by metabolism and not by liver perfusion [26]. Several studies have described a decrease in the activity and/or protein expression of CYPs in livers of patients with liver cirrhosis [43,44,45]. In these studies, the CYP3A4 protein expression was reduced by 70–80% [43, 44] and the activity in isolated microsomes by 40–60% [43, 45]. In vivo studies assessing the PK of the CYP3A model substrate midazolam after intravenous administration showed a reduction in midazolam clearance by 50–70% in patients with Child–Pugh B and C liver cirrhosis [34, 46]. These data match with the observed decrease in the formation of M1 and M3 in patients with moderate cirrhosis compared with healthy controls, whose AUC0–48h were decreased by 49% and 35%, respectively. This is also shown by the MR of M1 and M3, which may be used as an estimate of CYP3A4 activity. The MR of M1 was approximately doubled in patients with mild hepatic impairment and tripled in patients with moderate hepatic impairment.
Overall, a single oral dose of 25 mg of daridorexant was well tolerated both by healthy control subjects and by patients with liver cirrhosis, while the safety profile was similar to that observed in other studies [14, 16]. Importantly, all AEs (e.g., somnolence) in the present study were reported by subjects with hepatic impairment, which might be explained by the increased susceptibility of patients with liver cirrhosis caused by the underlying liver disease and associated encephalopathic effects [19, 47].
5 Conclusions
The PK of unbound daridorexant shows expected alterations in patients with moderate liver cirrhosis compared with control subjects: a reduced CL/F, explained mainly by impaired hepatic CYP3A4 activity, an increase in AUC0–inf, and impaired elimination velocity, but no increase in Cmax. No such changes were observed in patients with mild cirrhosis. No safety issue of concern was detected following administration of 25 mg of daridorexant in subjects with mild and moderate hepatic impairment as well as in healthy subjects. All AEs were transient and of mild/moderate intensity. Based on these data, a dose adjustment is not required in subjects with mild hepatic impairment. In subjects with moderate hepatic impairment, a dose of 25 mg should not be exceeded, considering the prolonged t½ of daridorexant. As patients with severe hepatic impairment were not studied, daridorexant should not be used in such patients. The study shows the importance of quantifying the free drug concentrations in the assessment of the PK of drugs with high plasma protein binding in patients with liver cirrhosis.
References
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(5):573–85.
Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8(3):171–81.
Treiber A, de Kanter R, Roch C, Gatfield J, Boss C, von Raumer M, et al. The use of physiology-based pharmacokinetic and pharmacodynamic modeling in the discovery of the dual orexin receptor antagonist ACT-541468. J Pharmacol Exp Ther. 2017;362(3):489–503.
Wang C, Wang Q, Ji B, Pan Y, Xu C, Cheng B, et al. The orexin/receptor system: molecular mechanism and therapeutic potential for neurological diseases. Front Mol Neurosci. 2018;11:220.
Muehlan C, Vaillant C, Zenklusen I, Kraehenbuehl S, Dingemanse J. Clinical pharmacology, efficacy, and safety of orexin receptor antagonists for the treatment of insomnia disorders. Expert Opin Drug Metab Toxicol. 2020;16(11):1063–78.
Mieda M, Sakurai T. Orexin (hypocretin) receptor agonists and antagonists for treatment of sleep disorders: rationale for development and current status. CNS Drugs. 2013;27(2):83–90.
Brisbare-Roch C, Dingemanse J, Koberstein R, Hoever P, Aissaoui H, Flores S, et al. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med. 2007;13(2):150–5.
Hoever P, Dorffner G, Beneš H, Penzel T, Danker-Hopfe H, Barbanoj MJ, et al. Orexin receptor antagonism, a new sleep-enabling paradigm: a proof-of-concept clinical trial. Clin Pharmacol Ther. 2012;91(6):975–85.
Dauvilliers Y, Zammit G, Fietze I, Mayleben D, Seboek Kinter D, Pain S, et al. Daridorexant, a new dual orexin receptor antagonist to treat insomnia disorder. Ann Neurol. 2020;87(3):347–56.
Muehlan C, Brooks S, Zuiker R, van Gerven J, Dingemanse J. Multiple-dose clinical pharmacology of ACT-541468, a novel dual orexin receptor antagonist, following repeated-dose morning and evening administration. Eur Neuropsychopharmacol. 2019;29(7):847–57.
Zammit G, Dauvilliers Y, Pain S, Sebök Kinter D, Mansour Y, Kunz D. Daridorexant, a new dual orexin receptor antagonist, in elderly subjects with insomnia disorder. Neurology. 2020;94(21):e2222–32.
Boof ML, Alatrach A, Ufer M, Dingemanse J. Interaction potential of the dual orexin receptor antagonist ACT-541468 with CYP3A4 and food: results from two interaction studies. Eur J Clin Pharmacol. 2019;75(2):195–205.
Zenklusen I, Muehlan C, Ulc I, Liska J, Dingemanse J. The dual orexin receptor antagonist daridorexant does not affect the pharmacokinetics of the BCRP substrate rosuvastatin. Clin Exp Pharmacol Physiol. 2020;47(11):1843–9.
Muehlan C, Zuiker R, Peeters P, Rowles R, Dingemanse J. Pharmacokinetics and pharmacodynamics of the dual orexin receptor antagonist daridorexant in Japanese and Caucasian subjects. J Clin Psychopharmacol. 2020;40(2):157–66.
Muehlan C, Heuberger J, Juif PE, Croft M, van Gerven J, Dingemanse J. Accelerated development of the dual orexin receptor antagonist ACT-541468: integration of a microtracer in a first-in-human study. Clin Pharmacol Ther. 2018;104(5):1022–9.
Muehlan C, Boehler M, Brooks S, Zuiker R, van Gerven J, Dingemanse J. Clinical pharmacology of the dual orexin receptor antagonist ACT-541468 in elderly subjects: exploration of pharmacokinetics, pharmacodynamics and tolerability following single-dose morning and repeated-dose evening administration. J Psychopharmacol. 2020;34(3):326–35.
Muehlan C, Fischer H, Zimmer D, Aissaoui H, Grimont J, Boss C, et al. Metabolism of the dual orexin receptor antagonist ACT-541468, based on microtracer/accelerator mass spectrometry. Curr Drug Metab. 2019;20(4):254–65.
Tapper EB, Henderson JB, Parikh ND, Ioannou GN, Lok AS. Incidence of and risk factors for hepatic encephalopathy in a population-based cohort of Americans with cirrhosis. Hepatol Commun. 2019;3(11):1510–9.
Córdoba J, Cabrera J, Lataif L, Penev P, Zee P, Blei AT. High prevalence of sleep disturbance in cirrhosis. Hepatology. 1998;27(2):339–45.
Martin Mateos R, Garcia de la Filia Molina I, Albillos A. Pre-surgical risk assessment in patients with cirrhosis. Acta Gastroenterol Belg. 2020;83(3):449–53.
Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60(8):646–9.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
Donzelli M, Derungs A, Serratore MG, Noppen C, Nezic L, Krahenbuhl S, et al. The basel cocktail for simultaneous phenotyping of human cytochrome P450 isoforms in plasma, saliva and dried blood spots. Clin Pharmacokinet. 2014;53(3):271–82.
Åkerstedt T, Gillberg M. Subjective and objective sleepiness in the active individual. Int J Neurosci. 1990;52(1–2):29–37.
Boss C, Gatfield J, Brotschi C, Heidmann B, Sifferlen T, von Raumer M, et al. The quest for the best dual orexin receptor antagonist (daridorexant) for the treatment of insomnia disorders. Chem Med Chem. 2020;15(23):2286–305.
Delco F, Tchambaz L, Schlienger R, Drewe J, Krahenbuhl S. Dose adjustment in patients with liver disease. Drug Saf. 2005;28(6):529–45.
Quigley EM. Gastrointestinal dysfunction in liver disease and portal hypertension: gut-liver interactions revisited. Dig Dis Sci. 1996;41(3):557–61.
Sadik R, Abrahamsson H, Björnsson E, Gunnarsdottir A, Stotzer PO. Etiology of portal hypertension may influence gastrointestinal transit. Scand J Gastroenterol. 2003;38(10):1039–44.
Thalheimer U, De Iorio F, Capra F, del Mar LM, Zuliani V, Ghidini V, et al. Altered intestinal function precedes the appearance of bacterial DNA in serum and ascites in patients with cirrhosis: a pilot study. Eur J Gastroenterol Hepatol. 2010;22(10):1228–34.
Isobe H, Sakai H, Satoh M, Sakamoto S, Nawata H. Delayed gastric emptying in patients with liver cirrhosis. Dig Dis Sci. 1994;39(5):983–7.
Pimpo MT, Frieri G, Saltarelli P, Ciccocioppo R, Aggio A, Marchetti G, et al. Effects of cisapride on abnormally prolonged endogastric alkalinity time and delayed gastric emptying in cirrhotic patients. Hepatogastroenterology. 1996;43(12):1678–84.
Usami A, Mizukami Y, Onji M. Abnormal gastric motility in liver cirrhosis: roles of secretin. Dig Dis Sci. 1998;43(11):2392–7.
Taegtmeyer AB, Haschke M, Tchambaz L, Buylaert M, Tschopl M, Beuers U, et al. A study of the relationship between serum bile acids and propranolol pharmacokinetics and pharmacodynamics in patients with liver cirrhosis and in healthy controls. PLoS ONE. 2014;9(6):e97885.
Albarmawi A, Czock D, Gauss A, Ehehalt R, Lorenzo Bermejo J, Burhenne J, et al. CYP3A activity in severe liver cirrhosis correlates with Child-Pugh and model for end-stage liver disease (MELD) scores. Br J Clin Pharmacol. 2014;77(1):160–9.
Xie F, Ding X, Zhang QY. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm Sin B. 2016;6(5):374–83.
Ceryak S, Bouscarel B, Fromm H. Comparative binding of bile acids to serum lipoproteins and albumin. J Lipid Res. 1993;34(10):1661–74.
Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147–61.
Barre J, Houin G, Rosenbaum J, Zini R, Dhumeaux D, Tillement JP. Decreased alpha 1-acid glycoprotein in liver cirrhosis: consequences for drug protein binding. Br J Clin Pharmacol. 1984;18(4):652–3.
Barre J, Mallat A, Rosenbaum J, Deforges L, Houin G, Dhumeaux D, et al. Pharmacokinetics of erythromycin in patients with severe cirrhosis: respective influence of decreased serum binding and impaired liver metabolic capacity. Br J Clin Pharmacol. 1987;23(6):753–7.
Knott C, Williams CP, Reynolds F. Phenytoin kinetics during pregnancy and the puerperium. Br J Obstet Gynaecol. 1986;93(10):1030–7.
Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1–8.
Klotz U, Avant GR, Hoyumpa A, Schenker S, Wilkinson GR. The effects of age and liver disease on the disposition and elimination of diazepam in adult man. J Clin Invest. 1975;55(2):347–59.
George J, Murray M, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology. 1995;21(1):120–8.
Prasad B, Bhatt DK, Johnson K, Chapa R, Chu X, Salphati L, et al. Abundance of phase 1 and 2 drug-metabolizing enzymes in alcoholic and hepatitis C cirrhotic livers: a quantitative targeted proteomics study. Drug Metab Dispos. 2018;46(7):943–52.
Yang LQ, Li SJ, Cao YF, Man XB, Yu WF, Wang HY, et al. Different alterations of cytochrome P450 3A4 isoform and its gene expression in livers of patients with chronic liver diseases. World J Gastroenterol. 2003;9(2):359–63.
MacGilchrist AJ, Birnie GG, Cook A, Scobie G, Murray T, Watkinson G, et al. Pharmacokinetics and pharmacodynamics of intravenous midazolam in patients with severe alcoholic cirrhosis. Gut. 1986;27(2):190–5.
McConnell JB, Curry SH, Davis M, Williams R. Clinical effects and metabolism of diazepam in patients with chronic liver disease. Clin Sci (Lond). 1982;63(1):75–80.
Acknowledgements
The authors thank the study team of the University Hospital Basel, Basel, Switzerland (Division of Clinical Pharmacology and Toxicology) with special thanks to the study nurses Claudia Bläsi and Joyce Jesus de Santos and to Beatrice Vetter for their valuable help during the preparation and conduct of the study and the study physicians Dr. Tanja Grandinetti, Dr. Florian Pfefferkorn, and Dr. Tim Bühler, Radka Štěpánová (Aixial s.r.o., Brno, Czech Republic) for statistical analysis of the clinical data, and Susanne Globig (Department of Preclinical Pharmacokinetics and Metabolism, Idorsia Pharmaceuticals Ltd, Allschwil, Switzerland) and Mark Enzler (Swiss BioQuant AG, Reinach, Switzerland) for the bioanalytical conduct. Last but not least, the authors thank the clinical research team, i.e., Alexandre Mathis, István Kerekes, Roberta Renai, Vincent Lemoine, Priska Kaufmann, and Pascale Gasser (Department of Clinical Pharmacology, Idorsia Pharmaceuticals Ltd, Allschwil, Switzerland).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was sponsored by Idorsia Pharmaceuticals Ltd, Allschwil, Switzerland.
Conflicts of Interest
Benjamin Berger, Jasper Dingemanse, Giancarlo Sabattini, Stéphane Delahaye, and Clemens Muehlan were full-time employees of Idorsia Pharmaceuticals Ltd during the conduct of the study. Jasper Dingemanse, Stéphane Delahaye, and Clemens Muehlan own stocks (options) of Idorsia Pharmaceuticals Ltd. Urs Duthaler and Stephan Krähenbühl were employees of the University Hospital Basel (Division of Clinical Pharmacology and Toxicology) and, in addition, Urs Duthaler was also employed by the University of Basel (Department of Biomedicine) during the conduct of the study. There are no other relationships or activities that could appear to have influenced the submitted work. The University Hospital Basel (Division of Clinical Pharmacology and Toxicology) received financial compensation for the clinical conduct.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in the study prior to any study-mandated procedure.
Consent for publication
Not available.
Availability of data and material
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Code availability
Not applicable.
Author contributions
JD and CM designed the study. UD and SK performed the assessments and collected the data. Data were analyzed by BB, JD, GS, SD, UD, CM, and SK. BB, JD, and SK wrote the manuscript. All authors reviewed and approved the final manuscript. The authors confirm that the principal investigator for this paper is SK and that he had direct clinical responsibility for the subjects.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Berger, B., Dingemanse, J., Sabattini, G. et al. Effect of Liver Cirrhosis on the Pharmacokinetics, Metabolism, and Tolerability of Daridorexant, A Novel Dual Orexin Receptor Antagonist. Clin Pharmacokinet 60, 1349–1360 (2021). https://doi.org/10.1007/s40262-021-01028-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01028-8