
Abstract
Purpose
ACT-541468 is a novel dual orexin receptor antagonist (DORA) under development for the treatment of insomnia. In vitro studies suggested a significant role of CYP3A4 in ACT-541468 metabolism and an impact on CYP3A4 activity.
Methods
Subsequently, two clinical cross-over studies investigated the victim (n = 14 healthy subjects) and perpetrator (n = 20) potential of 25 mg ACT-541468 with respect to CYP3A4. The effect of food intake on the pharmacokinetics of ACT-541468 was also investigated.
Results
Moderate CYP3A4 inhibition by diltiazem (240 mg/day) increased the Cmax and AUC0–∞ of ACT-541468 by 1.4-fold (90% confidence interval (CI): 1.2–1.6) and 2.4-fold (90% CI: 2.0–2.8), respectively, and prolonged t½ by 80% (90% CI: 60–90) without affecting tmax. Single- and multiple-dose administration of 25 mg ACT-541468 had no impact on the pharmacokinetics of the sensitive substrate midazolam and its main metabolite 1-hydroxy midazolam indicated by 90% CI of the geometric mean ratios of Cmax and AUC within bioequivalence criteria and by an unchanged tmax. After a high-fat high-calorie breakfast, the pharmacokinetic profile of 25 mg ACT-541468 showed a decrease of Cmax by 24% (90% CI: 17–31) and a delay of tmax by approximately 2 h (90% CI: 1.4–2.4), whereas t½ and AUC0–24 remained essentially unchanged. ACT-541468 given alone or in combination with diltiazem, midazolam, or food was safe and well tolerated.
Conclusions
Overall, ACT-541468 has been determined as CYP3A4 substrate but without any perpetrator drug–drug interaction potential regarding CYP3A4 in humans. Food affected ACT-541468 absorption without modifying overall exposure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The orexin system, discovered in 1998, involves two neuropeptides orexin A and orexin B that are synthesized in the lateral hypothalamic area which projects to various regions in the brain and that binds two G protein-coupled receptors orexin-1 (OX1) and orexin-2 (OX2) [1, 2]. This system plays a central role in the regulation of arousal and sleep–wake balance [3,4,5]. To date, a number of dual orexin receptor antagonists (DORAs) have entered clinical development for the treatment of sleep disorders: almorexant [6, 7], ACT-462206 [8], SB-649868 [9], lemborexant [10], filorexant [11], and suvorexant [12]. In 2014, suvorexant was granted market authorization in the US and Japan (Belsomra®) for the treatment of insomnia.
ACT-541468 ((S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5 methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone) is a novel DORA [13] that is currently developed for the treatment of insomnia. The chemical structure is provided in the supplemental Fig. S1. In the first-in-human study, ACT-541468 was characterized by quick absorption and elimination, with median time to reach maximum concentration (tmax) of 0.8–2.8 h and geometric mean elimination half-life (t½) of 5.9 to 8.8 h. Dose-related sedative effects were observed indicating pharmacokinetic (PK) and pharmacodynamic (PD) profiles suitable for a sleep-promoting compound [14].
The role of cytochrome P450 (CYP) 3A4 in the metabolism of DORAs has been extensively evaluated in preclinical and clinical development stages and it was shown that this CYP is the main enzyme involved in the metabolism of suvorexant [15], almorexant [16], and SB-649868 [17]. In vitro studies were performed to quantify the role of CYP3A4 in ACT-541468 metabolism and to assess its inhibitory and inductive effects on CYP3A4 activity (data on file). Based on these data, > 80% of ACT-541468 clearance was predicted to be mediated by CYP3A4. In human liver microsomes, CYP3A4 activity (i.e., midazolam and testosterone hydroxylation) was inhibited with an IC50 of approximately 10 μM. Therefore, two clinical drug–drug interaction (DDI) studies have been conducted to assess the victim and perpetrator DDI potential of ACT-541468 with respect to CYP3A4. In addition, the effect of a high-fat high-calorie breakfast on the PK of ACT-541468 was assessed to better define clinical instructions of use in the pivotal studies.
Methods
Each study was approved by the German health authority (BfArM) and by the Ethics committee (Ethikkommission II, Bad Segeberg, Germany). They were both conducted in accordance with the Declaration of Helsinki principles, International Council for Harmonization Good Clinical Practice guidelines, and applicable regulations and laws. All subjects provided written informed consent prior to any screening procedures.
Study designs
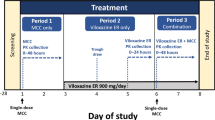
Study 1 (Fig. 1a) was a single-center, open-label, randomized, cross-over study in 14 subjects (7 subjects per sequence) to investigate the effect of the moderate CYP3A4 inhibitor diltiazem on the PK of ACT-541468. Each subject received a single oral dose of 25 mg ACT-541468 under fasting conditions alone (Treatment A) or on Day 4 of a 7-day multiple, oral dose regimen of 240 mg diltiazem once daily for 7 days (Treatment B), i.e., at steady-state exposure of diltiazem [18, 19].

Schematic presentation of the clinical study design for (a) study 1 (CYP3A4 victim potential of ACT-541468) and (b) study 2 (CYP3A4 perpetrator potential of ACT-541468)
Study 2 (Fig. 1b) was a single-center, open-label, fixed-sequence study in 20 subjects to investigate the effect of ACT-541468 on the PK of the CYP3A4 index substrate midazolam [20] and its main metabolite 1-hydroxy midazolam (1-OH midazolam) as well as the effect of a high-fat high-calorie breakfast on the PK of ACT-541468. Each subject received a single, oral dose of 2 mg midazolam under fasting conditions alone (Treatment A) and in combination with single (Treatment B) and multiple (Treatment D) oral doses of 25 mg ACT-541468 once daily for 5 days. For the assessment of food effect on the PK of ACT-541468, subjects were given a high-fat high-calorie breakfast 30 min prior to the administration of a single dose of 25 mg ACT-541468 as per Food and Drug Administration (FDA) guidance [21] and European Medicines Agency (EMA) guidelines [22] during Treatment C. ACT-541468 PK parameters under fasting conditions were assessed during Treatment B.
The safety and tolerability of ACT-541468 administered alone or in combination with either diltiazem or midazolam was also assessed.
For these two bio-comparison studies, the within-subject standard deviation (SDw) on log scale observed for area under the plasma concentration–time curve from zero to infinity (AUC0–∞, study 1) or AUC from 0 to 24 h after administration (AUC0–24, study 2) and maximum observed plasma concentration (Cmax) were estimated from previous studies for ACT-541468 [14] and for midazolam/1-OH midazolam [23]. For ACT-541468, SDw was 0.30 for AUC and 0.15 for Cmax. For midazolam and 1-OH midazolam, SDw was 0.44 and 0.41, respectively, for AUC and 0.36 and 0.44, respectively, for Cmax. In study 1, it was estimated that, with a sample size of 12 subjects, the 90% confidence interval (CI) of the geometric mean ratio (GMR) “ACT 541468 + diltiazem/ACT-541468” would be 0.86–1.17 for AUC0–∞ and 0.92–1.08 for Cmax. In study 2, corresponding data for “midazolam + ACT-541468/midazolam” were, with a sample size of 16 subjects, 0.66–1.52 for AUC0–24 and 0.71–1.41 for Cmax of midazolam and 0.68–1.52 for AUC0–24 and 0.66–1.52 for Cmax of 1-OH midazolam.
To compensate for any potential dropouts, 14 subjects (study 1) and 20 subjects (study 2) were enrolled to achieve a minimum of 12 and 16 subjects, respectively, completing the studies per protocol.
Subjects
Non-smoking, healthy male adults 18–45 years old with a body mass index (BMI) of 18–28 kg/m2 (study 1) or 18–30 kg/m2 (study 2) were eligible for study entry. To be eligible, the results of the self-administered modified Swiss Narcolepsy Scale questionnaire [24] had to show no signs of narcolepsy/cataplexy.
Consumption of nicotine, alcohol, or food and beverages that could influence CYP3A activity (e.g., grapefruit or xanthine-containing beverages) was forbidden. Concomitant therapy was prohibited unless required for the treatment of an adverse event (AE).
Blood sampling and bioanalysis of ACT-541468, midazolam, and 1-OH midazolam concentrations in plasma
Serial blood samples were collected in EDTA tubes for 96 h (study 1) and for 24 h (study 2) for the determination of ACT-541468 (study 1 and 2) and midazolam and 1-OH midazolam (study 2) plasma concentrations. In study 2, pre-dose samples from day 5 to day 7 were also collected to assess the trough plasma concentrations (Ctrough) of ACT-541468. After centrifugation, plasma was transferred to polypropylene tubes and stored at − 20 °C or below.
Concentrations of ACT-541468, midazolam, and 1-OH midazolam in plasma were measured using previously described validated liquid chromatography methods with tandem mass spectrometry (LC-MS/MS) [14, 25]. The lower limit of quantification (LLOQ) was 0.5 ng/mL for ACT-541468 and 0.1 ng/mL for both midazolam and 1-OH midazolam. For ACT-541468, inter-batch precision expressed as coefficient of variation (%CV) was ≤ 5.4% (study 1) and ≤ 4.4% (study 2), whereas the inter-batch accuracy expressed as relative deviation from nominal value (%RD) ranged from − 6.2% to − 1.9% (study 1) and from − 0.8 to 3.2% (study 2). For midazolam and 1-OH midazolam, %CV was ≤ 6.0% and ≤ 6.6%, respectively, whereas %RD ranged from − 1.9 to 2.3% and from − 3.7 to 0.3%, respectively.
Pharmacokinetic calculations
The PK parameters of ACT-541468 in both studies as well as of midazolam and 1-OH midazolam (study 2) were obtained by non-compartmental analysis using Phoenix WinNonlin (version 6.4; Pharsight Corporation, Mountain View, CA, USA). The measured individual plasma concentrations of each analyte were used to directly obtain Cmax and tmax. AUC values were calculated according to the linear trapezoidal rule, using the measured concentration values above the LLOQ, without any weighting. The t½ of all analytes was calculated as follows: t½ = ln(2)/λz, where λz represents the terminal elimination rate constant. Assessment of steady-state concentrations of ACT-541468 (study 2) was based on Ctrough data.
Statistical analysis of pharmacokinetic variables
The PK analysis was based on the per-protocol analysis set, which included all subjects that completed the dosing as per protocol. PK variables were analyzed providing geometric means and corresponding 95% CI for AUC, Cmax, and t½ whereas the median and range were used for tmax.
The effects of diltiazem and food on AUC, Cmax, and t½ of ACT-541468 were explored using the GMR and 90% CIs of “ACT-541468 + diltiazem” (study 1, Treatment B) or “ACT-541468 + food” (study 2, Treatment C) with ACT-541468 alone as reference treatment.
Similarly, the effect of ACT-541468 on AUC, Cmax, and t½ of midazolam and 1-OH midazolam was explored using the GMR and 90% CIs of “midazolam + single-dose ACT-541468” (study 2, Treatment B) or “midazolam + multiple-dose ACT-541468” (study 2, Treatment D) with midazolam alone as reference treatment.
The log-transformed values were analyzed by mixed-effect models including treatment, sequence (study 1 only), and period as fixed effects and subject as random effect. Differences between treatments for tmax were explored using the Wilcoxon signed-rank test providing the median differences and corresponding 90% CI.
All statistical analyses were performed using SAS® version 9.3 (SAS Institute, Cary, NC, USA).
Safety and tolerability
Safety and tolerability were assessed based on vital sign, 12-lead electrocardiogram (ECG), physical examination, clinical chemistry, hematology, and AE data. The all-treated analysis set, which included all subjects who received at least one dose of treatment, was used for the safety analysis.
Results
Study population
A total of 14 (study 1) and 20 (study 2) subjects were enrolled in this study and were included in the all-treated analysis set. All subjects completed the studies as per protocol except for one subject in study 1 who discontinued the treatment because he did not come to the study site to receive diltiazem on Day 2 and was thus excluded from the per-protocol analysis set. In both studies, all subjects were Caucasian except one per study who was Asian. Demographic characteristics (age and BMI) were similar across the Treatment sequences in study 1 and between both studies (Table 1). None of the subjects deviated from any inclusion/exclusion criteria.
Pharmacokinetic results
Effect of the moderate CYP3A4 inhibitor diltiazem on the PK of ACT-541468
Concomitant administration of 240 mg diltiazem once daily increased Cmax of ACT-541468 by 1.4-fold and AUC0–∞ by 2.4-fold (Figure 2, Table 2), indicating ACT-541468 as sensitive substrate of CYP3A4 in humans according to FDA guidance [26]. ACT-541468 t½ was prolonged by 5.4 h whereas tmax was shortened by 15 min (Fig. 2, Table 2).

Arithmetic mean (± SD) plasma concentration–time profile of ACT-541468 after administration of ACT-541468 alone and concomitantly with diltiazem (n = 13) on a linear and semi-logarithmic scale
Effect of ACT-541468 on the PK of the CYP3A4 index substrate midazolam and its metabolite 1-OH midazolam
Steady-state plasma concentrations of ACT-541468 were reached on the second day of multiple-dose administration based on Ctrough data (Fig. S2a). The PK profile and parameters of ACT-541468 were similar after single- and multiple-dose administration (Fig. S2b, Table 3) indicating no relevant accumulation.
After single-dose ACT-541468 administration, geometric mean Cmax and AUC0–24 of midazolam were increased by 20 and 11%, respectively (Fig. 3a and S3a, Table 3), whereas they were increased by only 5% for 1-OH midazolam (Fig. 3b and S3b, Table 3). As for Cmax and AUC0–24, t½ and tmax of both analytes were essentially unchanged when compared to midazolam given alone (Fig. 3a, b, S3a, and S3b, Table 3).

Arithmetic mean (± SD) plasma concentration–time profile of (a) midazolam and (b) 1-OH midazolam after administration of midazolam alone, 1 h after single-dose ACT-541468, and after multiple-dose 25 mg ACT-541468 from time 0 to 6 h post-dose on a linear and semi-logarithmic scale (n = 20)
After multiple-dose ACT-541468 administration, geometric mean Cmax and AUC0–24 of midazolam were decreased by 6.4 and 2.5%, respectively (Fig. 3a and S3a, Table 3), whereas 1-OH midazolam Cmax was only decreased by 1.8% and AUC0–24 remained unchanged (Fig. 4b and S3b, Table 4). Accordingly, after single-dose ACT-541468 administration, t½ and tmax of both analytes were essentially unchanged when compared to midazolam given alone (Fig. 3a, b, S3a, and S3b, Table 3).
Overall, the PK of midazolam and 1-OH midazolam were essentially unaffected by single- and multiple-dose administration of 25 mg ACT-541468.
Effect of food on the PK of ACT-541468
In the presence of food, Cmax of ACT-541468 was decreased by 24% and tmax was delayed by approximately 2 h, whereas t½ and AUC0–24 remained essentially unchanged (increase of 2.6 and 3.2%, respectively) (Fig. 4, Table 4).

Arithmetic mean (± SD) plasma concentration–time profile of ACT-541468 after administration of ACT-541468 under fasting and fed conditions (n = 20) on a linear and semi-logarithmic scale
Safety and tolerability
Single oral doses of 25 mg ACT-541468 alone or in combination with multiple oral doses of 240 mg diltiazem, high-fat high-calorie breakfast, and single oral dose of 2 mg midazolam were safe and tolerated. There were no clinically relevant effects on vital sign, ECG, or laboratory variables. None of the AEs were serious or led to discontinuation from the study. All treatment-emergent AEs were transient and resolved without sequelae before the end of the studies.
In both studies, the most frequently reported AEs were somnolence and headache. Somnolence was reported by each subject exposed to ACT-541468 alone or in combination with either diltiazem, food, or midazolam. These AEs of somnolence were rated as mild (33.3%), moderate (63.2%), or severe (3.5%) based on pooled analysis from both studies. The second most common AE was headache reported by 50% of the subjects in study 1 mostly after the administration of diltiazem alone and by 25% of the subjects in study 2, exclusively after the administration of midazolam alone. These AEs were mild (71.4%) or moderate (28.6%) in intensity.
Discussion
In line with the in vitro data suggesting ACT-541468 as CYP3A4 substrate, the first clinical study shows that the single-dose PK of ACT-541468 are modulated by the moderate CYP3A4 inhibitor diltiazem. However, the second clinical study shows that ACT-541468 itself does not modulate the PK of the CYP3A4 index substrate midazolam despite some inhibition/induction potential identified in vitro. This study also shows that a high-fat high-calorie breakfast has no impact on the overall exposure to ACT-541468 but modulates Cmax and tmax to some extent.
Both exploratory bio-comparison studies were conducted in line with the applicable guidelines in terms of design and choice of interacting compounds/meal content for interaction with food [20,21,22]. ACT-541468 was administered at a dose of 25 mg (single and multiple) since it is considered as potential therapeutically relevant dose for the treatment of insomnia triggering the desired PD effects with the appropriate onset and duration of action [14]. Both studies included only male subjects to have a more homogenous population.
ACT-541468 victim potential in the context of moderate CYP3A4 inhibition
To evaluate the relevance of the effect of CYP3A4 on ACT-541468 metabolism in humans, the moderate CYP3A4 inhibitor diltiazem was administered once daily at the therapeutic dose of 240 mg for 7 days to reach steady-state conditions [18] and maintain continuous inhibition during the elimination phase of ACT-541468 [27, 28]. In previous DDI studies, steady-state conditions were attained on the third day of diltiazem administration [28] and maximal CYP3A4 inhibition was obtained after 2 days of administration [18]. The same dose of diltiazem was used for the evaluation of suvorexant DDI potential with a moderate CYP3A4 inhibitor [29]. After concomitant administration with diltiazem, Cmax, AUC0–∞, and t½ of ACT-541468 were increased by 1.4-, 2.4-, and 1.8-fold, respectively. The increase of Cmax and AUC as well as the longer t½ indicates that both absorption and elimination are affected by moderate CYP3A4 inhibition. The 2.4-fold AUC0–∞ increase of ACT-541468 upon co-administration of the moderate CYP3A4 inhibitor diltiazem is contained in the 2- to 5-fold range and hence, ACT-541468 may be considered as “sensitive” CYP3A4 substrate according to FDA guidance in 2012 [26]/EMA guidelines [22] on clinical DDI studies. The larger AUC0–∞ of ACT-541468 in the presence of diltiazem did not translate into an increased intensity classification of somnolence when compared to ACT-541468 given alone. However, the longer t½ of ACT-541468 could translate in a longer duration of somnolence.
Clinical DDI studies with other DORAs are restricted to suvorexant [29] and almorexant [16]. Here, the AUC of suvorexant (20 mg) and almorexant (100 mg) was increased by 2.1- and 3.7-fold, respectively, upon multiple-dose administration of 240 mg or 300 mg diltiazem, respectively. Similarly, Cmax was increased by 1.2- and 3.2-fold for suvorexant and almorexant, respectively.
At the time of study conduct, both FDA guidance and EMA guidelines recommended the use of a strong CYP3A4 inhibitor (so-called perpetrator index in the new FDA draft guidance [20]) to assess the DDI potential with CYP3A4 [22, 26]. However, this study was conducted with a moderate CYP3A4 inhibitor since concomitant administration of ACT-541468 at a therapeutically relevant dose with a strong CYP3A4 inhibitor may have led to an AUC exceeding that of the highest dose of 200 mg investigated in the single-ascending dose study [14] as preliminary model-based predictions suggested a more than 7-fold increase of ACT-541468 AUC in the presence of a strong inhibitor such as ketoconazole. Even though the study results still allow to conclude that ACT-541468 is a sensitive CYP3A4 substrate, the lacking evaluation of a worst-case DDI scenario has to be considered as study limitation. However, physiology-based PK modeling has recently gained increasing acceptance by the US and European authorities and may be used here to predict changes in drug exposure related to inhibition or induction of drug-metabolizing enzymes [20, 22].
Perpetrator potential of ACT-541468 on CYP3A4 metabolism
In study 2, the well-established CYP3A4 index substrate midazolam was used as interacting treatment and administered at a low dose of 2 mg to ensure an acceptable tolerability when given in combination with ACT-541468 given that both compounds exhibit sedative effects, while still allowing for appropriate evaluation of the PK endpoints [30], and for comparison with other DORAs [23, 29]. Midazolam is mainly metabolized by CYP3A4 in the liver and gut wall [31, 32] to form the major active metabolite 1-OH midazolam. This metabolite is thus a good “biomarker” of any potential modification of CYP3A4 metabolism. The study was designed to evaluate both the CYP3A4 inhibition and induction potential of ACT-541468 in a single study as previously described [33]. Whereas enzyme induction requires a prolonged administration of a perpetrator [34,35,36], enzyme inhibition/inactivation, even if not maximal, is observed after a single-dose administration of the inhibitor [37, 38]. Thus, in order to adequately assess the CYP3A4 inhibition potential, midazolam was given at the previously reported tmax [14] of ACT-541468, i.e., 1 h after ACT-541468 administration, while the induction potential (i.e., net effect of induction/inhibition) was investigated during steady-state conditions [16, 25] of ACT-541468. These were already reached after the second day of administration in line with the previously reported t½ of 6.1 h [14] (Fig. S2a).
A lack of perpetrator DDI potential with respect to CYP3A4 has been determined for ACT-541468 given that the 90% CIs of the GMR were all contained in the 0.8–1.25 equivalence boundaries and median tmax of midazolam and 1-OH midazolam were not different in the absence or presence of ACT-541468. These results seem contradictory to the ones obtained in vitro where ACT-541468 was shown to be both an inhibitor and inducer of CYP3A4 (data on file). However, taking into consideration a mean plasma Cmax after single-dose/multiple-dose administration in study 2 of approximately 750 ng/mL (i.e., equivalent to 1.54 μM) and the very high plasma protein binding [13], the levels of unbound ACT-541468 in the present clinical study are at nanomolar level which is far below the required concentration for CYP3A4 inhibition and induction in the liver, i.e., at the micromolar level. Considering a gastric volume < 50 mL under fasting conditions [39] to which are added the 240 mL of water given during administration, the maximum concentration of an oral dose of 25 mg ACT-541468 at the level of the gut would be 0.18 μM, which is also not sufficient to inhibit/induce CYP3A4. The similarity of ACT-541468 plasma exposure observed between Day 1 and Day 5 (Fig. S2b) further supports the lack of effect of ACT-541468 on CYP3A4 in vivo knowing from the study with diltiazem that ACT-541468 PK are modulated by CYP3A4.
With respect to suvorexant, a supra-therapeutic multiple dose of 80 mg increased the AUC of midazolam by 1.5-fold [29] indicating suvorexant as weak inhibitor of CYP3A4.
Interaction of ACT-541468 with food
The effect of food on ACT-541468 PK parameters was evaluated using a high-fat breakfast to ensure a “worst-case scenario” with relation to food intake, i.e., a meal content that induces a maximum effect on the gastrointestinal tract, as per FDA guidance [21] and EMA guidelines [22].
Concomitant administration of a high-fat breakfast had no impact on the extent of ACT-541468 exposure given that t½ and AUC remained unchanged. However, a decreased Cmax and delayed tmax has been observed when ACT-541468 was administered together with food. Hence, peak effects of ACT-541468 could principally be decreased and delayed. The underlying mechanisms of such effects were not investigated in the study but decreased Cmax and delayed tmax usually result from a slower gastric emptying rate and/or increased gastric pH induced by food intake [40]. In case of suvorexant, a similar delayed tmax (1.5 h) after high-fat breakfast intake has been reported, but Cmax nor AUC were affected [29].
One limitation of the food effect assessment relates to the use of combined administration of ACT-541468 and midazolam as fasted reference treatment and previously reported midazolam-mediated delay of gastric emptying in mice [41]. However, this is not considered to have introduced major bias given that a much lower dose has been used in our study (2 mg orally, i.e., approximately 0.025 mg/kg) than in mice (25/50 mg/kg intraperitoneally). Moreover, no AEs indicative of delayed gastric emptying were reported.
While food–drug interactions can result in safety concerns when bioavailability is increased (i.e., higher Cmax), this appears not the case with ACT-541468 given the decrease of Cmax and the unchanged overall exposure.
Conclusion
In conclusion, these two clinical studies showed that ACT-541468 may be a sensitive CYP3A4 substrate, but is not a perpetrator of CYP3A4-mediated DDIs. Upon concomitant administration of a high-fat breakfast, overall exposure to ACT-541468 was unchanged, while Cmax was decreased by approximately 25% and tmax was delayed by approximately 2 h. Overall, these results support the further development of this new DORA for the treatment of patients suffering from insomnia.
References
de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A 95(1):322–327
Sakurai T (2007) The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci 8(3):171–181. https://doi.org/10.1038/nrn2092
Bonnavion P, de Lecea L (2010) Hypocretins in the control of sleep and wakefulness. Curr Neurol Neurosci Rep 10(3):174–179. https://doi.org/10.1007/s11910-010-0101-y
Inutsuka A, Yamanaka A (2013) The physiological role of orexin/hypocretin neurons in the regulation of sleep/wakefulness and neuroendocrine functions. Front Endocrinol (Lausanne) 4:18. https://doi.org/10.3389/fendo.2013.00018
Sakurai T, Mieda M, Tsujino N (2010) The orexin system: roles in sleep/wake regulation. Ann N Y Acad Sci 1200:149–161. https://doi.org/10.1111/j.1749-6632.2010.05513.x
Brisbare-Roch C, Dingemanse J, Koberstein R, Hoever P, Aissaoui H, Flores S, Mueller C, Nayler O, van Gerven J, de Haas SL, Hess P, Qiu C, Buchmann S, Scherz M, Weller T, Fischli W, Clozel M, Jenck F (2007) Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med 13(2):150–155. https://doi.org/10.1038/nm1544
Hoever P, Dorffner G, Benes H, Penzel T, Danker-Hopfe H, Barbanoj MJ, Pillar G, Saletu B, Polo O, Kunz D, Zeitlhofer J, Berg S, Partinen M, Bassetti CL, Hogl B, Ebrahim IO, Holsboer-Trachsler E, Bengtsson H, Peker Y, Hemmeter UM, Chiossi E, Hajak G, Dingemanse J (2012) Orexin receptor antagonism, a new sleep-enabling paradigm: a proof-of-concept clinical trial. Clin Pharmacol Ther 91(6):975–985. https://doi.org/10.1038/clpt.2011.370
Hoch M, van Gorsel H, van Gerven J, Dingemanse J (2014) Entry-into-humans study with ACT-462206, a novel dual orexin receptor antagonist, comparing its pharmacodynamics with almorexant. J Clin Pharmacol 54(9):979–986. https://doi.org/10.1002/jcph.297
Bettica P, Nucci G, Pyke C, Squassante L, Zamuner S, Ratti E, Gomeni R, Alexander R (2012) Phase I studies on the safety, tolerability, pharmacokinetics and pharmacodynamics of SB-649868, a novel dual orexin receptor antagonist. J Psychopharmacol 26(8):1058–1070. https://doi.org/10.1177/0269881111408954
Yoshida Y, Naoe Y, Terauchi T, Ozaki F, Doko T, Takemura A, Tanaka T, Sorimachi K, Beuckmann CT, Suzuki M, Ueno T, Ozaki S, Yonaga M (2015) Discovery of (1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluor opyridin-2-yl)cyclopropanecarboxamide (E2006): a potent and efficacious oral orexin receptor antagonist. J Med Chem 58(11):4648–4664. https://doi.org/10.1021/acs.jmedchem.5b00217
Connor KM, Mahoney E, Jackson S, Hutzelmann J, Zhao X, Jia N, Snyder E, Snavely D, Michelson D, Roth T, Herring WJ (2016) A phase II dose-ranging study evaluating the efficacy and safety of the orexin receptor antagonist filorexant (MK-6096) in patients with primary insomnia. Int J Neuropsychopharmacol 19(8):pyw022. https://doi.org/10.1093/ijnp/pyw022
Winrow CJ, Renger JJ (2014) Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol 171(2):283–293. https://doi.org/10.1111/bph.12261
Treiber A, de Kanter R, Roch C, Gatfield J, Boss C, von Raumer M, Schindelholz B, Muehlan C, van Gerven J, Jenck F (2017) The use of physiology-based pharmacokinetic and pharmacodynamic modeling in the discovery of the dual orexin receptor antagonist ACT-541468. J Pharmacol Exp Ther 362(3):489–503. https://doi.org/10.1124/jpet.117.241596
Muehlan C, Heuberger J, Juif PE, Croft M, van Gerven J, Dingemanse J (2018) Accelerated development of the dual orexin receptor antagonist ACT-541468: integration of a microtracer in a first-in-human study. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.1046
Cui D, Cabalu T, Yee KL, Small J, Li X, Liu B, Maciolek C, Smith S, Liu W, McCrea JB, Prueksaritanont T (2016) In vitro and in vivo characterisation of the metabolism and disposition of suvorexant in humans. Xenobiotica 46(10):882–895. https://doi.org/10.3109/00498254.2015.1129565
Dingemanse J, Cruz HG, Gehin M, Hoever P (2014) Pharmacokinetic interactions between the orexin receptor antagonist almorexant and the CYP3A4 inhibitors ketoconazole and diltiazem. J Pharm Sci 103(5):1548–1556. https://doi.org/10.1002/jps.23916
Renzulli C, Nash M, Wright M, Thomas S, Zamuner S, Pellegatti M, Bettica P, Boyle G (2011) Disposition and metabolism of [14C]SB-649868, an orexin 1 and 2 receptor antagonist, in humans. Drug Metab Dispos 39(2):215–227. https://doi.org/10.1124/dmd.110.035386
Friedman EJ, Fraser IP, Wang YH, Bergman AJ, Li CC, Larson PJ, Chodakewitz J, Wagner JA, Stoch SA (2011) Effect of different durations and formulations of diltiazem on the single-dose pharmacokinetics of midazolam: how long do we go? J Clin Pharmacol 51(11):1561–1570. https://doi.org/10.1177/0091270010387141
Bui K, Zhou D, Sostek M, She F, Al-Huniti N (2016) Effects of CYP3A modulators on the pharmacokinetics of naloxegol. J Clin Pharmacol 56(8):1019–1027. https://doi.org/10.1002/jcph.693
FDA Draft Guidance for Industry Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications (October 2017)
FDA Guidance for Industry Food-Effect Bioavailability and Fed Bioequivalence Studies. (December 2002)
Committee for Human Medicinal Products Guideline on the investigation of drug interactions. (June 2012). CPMP/EWP/560/95/Rev. 1 Corr. 2
Hoch M, Hoever P, Alessi F, Theodor R, Dingemanse J (2013) Pharmacokinetic interactions of almorexant with midazolam and simvastatin, two CYP3A4 model substrates, in healthy male subjects. Eur J Clin Pharmacol 69(3):523–532. https://doi.org/10.1007/s00228-012-1403-6
Sturzenegger C, Bassetti CL (2004) The clinical spectrum of narcolepsy with cataplexy: a reappraisal. J Sleep Res 13(4):395–406. https://doi.org/10.1111/j.1365-2869.2004.00422.x
Juif PE, Boehler M, Donazzolo Y, Bruderer S, Dingemanse J (2017) A pharmacokinetic drug–drug interaction study between selexipag and midazolam, a CYP3A4 substrate, in healthy male subjects. Eur J Clin Pharmacol 73(9):1121–1128. https://doi.org/10.1007/s00228-017-2282-7
FDA Draft Guidance for Industry Drug Interaction Studies—Study Design, Data Analysis, and Implications for Dosing and Labeling. (February 2012)
Dingemanse J, Nicolas L (2013) Drug–drug interaction study of ACT-178882, a new renin inhibitor, and diltiazem in healthy subjects. Clin Drug Investig 33(3):207–213. https://doi.org/10.1007/s40261-013-0056-2
Dingemanse J, Nicolas LB, van Bortel L (2013) Effect of multiple-dose diltiazem on the pharmacokinetics of the renin inhibitor ACT-077825. Clin Pharmacol Drug Dev (2, 2):113–119. https://doi.org/10.1002/cpdd.21
Merck (2014) Belsomra US Package Insert
McCrea J, Prueksaritanont T, Gertz BJ, Carides A, Gillen L, Antonello S, Brucker MJ, Miller-Stein C, Osborne B, Waldman S (1999) Concurrent administration of the erythromycin breath test (EBT) and oral midazolam as in vivo probes for CYP3A activity. J Clin Pharmacol 39(12):1212–1220. https://doi.org/10.1177/00912709922012015
Gorski JC, Hall SD, Jones DR, VandenBranden M, Wrighton SA (1994) Regioselective biotransformation of midazolam by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem Pharmacol 47(9):1643–1653. https://doi.org/10.1016/0006-2952(94)90543-6
Thummel KE, O'Shea D, Paine MF, Shen DD, Kunze KL, Perkins JD, Wilkinson GR (1996) Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther 59(5):491–502. https://doi.org/10.1016/S0009-9236(96)90177-0
Dutreix C, Munarini F, Lorenzo S, Roesel J, Wang Y (2013) Investigation into CYP3A4-mediated drug–drug interactions on midostaurin in healthy volunteers. Cancer Chemother Pharmacol 72(6):1223–1234. https://doi.org/10.1007/s00280-013-2287-6
Branch RA, Adedoyin A, Frye RF, Wilson JW, Romkes M (2000) In vivo modulation of CYP enzymes by quinidine and rifampin. Clin Pharmacol Ther 68(4):401–411. https://doi.org/10.1067/mcp.2000.110561
Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT (2003) Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet 42(9):819–850. https://doi.org/10.2165/00003088-200342090-00003
Zhou SF (2008) Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab 9(4):310–322
Bottiger Y, Sawe J, Brattstrom C, Tollemar J, Burke JT, Hass G, Zimmerman JJ (2001) Pharmacokinetic interaction between single oral doses of diltiazem and sirolimus in healthy volunteers. Clin Pharmacol Ther 69(1):32–40
Dhuria S, Einolf H, Mangold J, Sen S, Gu H, Wang L, Cameron S (2013) Time-dependent inhibition and induction of human cytochrome P4503A4/5 by an oral IAP antagonist, LCL161, in vitro and in vivo in healthy subjects. J Clin Pharmacol 53(6):642–653. https://doi.org/10.1002/jcph.79
Sjogren E, Abrahamsson B, Augustijns P, Becker D, Bolger MB, Brewster M, Brouwers J, Flanagan T, Harwood M, Heinen C, Holm R, Juretschke HP, Kubbinga M, Lindahl A, Lukacova V, Munster U, Neuhoff S, Nguyen MA, Peer A, Reppas C, Hodjegan AR, Tannergren C, Weitschies W, Wilson C, Zane P, Lennernas H, Langguth P (2014) In vivo methods for drug absorption—comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur J Pharm Sci 57:99–151. https://doi.org/10.1016/j.ejps.2014.02.010
Singh BN (1999) Effects of food on clinical pharmacokinetics. Clin Pharmacokinet 37(3):213–255. https://doi.org/10.2165/00003088-199937030-00003
Inada T, Asai T, Yamada M, Shingu K (2004) Propofol and midazolam inhibit gastric emptying and gastrointestinal transit in mice. Anesth Analg 99(4):1102–1106, table of contents. https://doi.org/10.1213/01.ANE.0000130852.53082.D5
Acknowledgments
The authors thank Dr. Atef Halabi and the study team at Clinical Research Services (CRS, Kiel, Germany) for the clinical conduct of both studies, Mariya Antonova and Radka Štěpánová (Aprova s.r.o., Brno, Czech Republic) for statistical analysis of clinical data, Susanne Globig (Department of Preclinical Pharmacokinetics and Metabolism, Idorsia Pharmaceuticals Ltd), and Lena Borkowski (ACC GmbH Analytical Clinical Concepts, Leidersbach, Germany) for the bioanalytical conduct of ACT-541468 and midazolam/1-OH midazolam, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Actelion Pharmaceuticals Ltd., the predecessor of Idorsia Pharmaceuticals Ltd., provided funding for this clinical study, as owner of ACT-541468. At the time of the study conduct or reporting, M.-L.B., M.U., and J.D. were full-time employees of Actelion Pharmaceuticals Ltd. They are now full-time employees of Idorsia Pharmaceuticals Ltd., the current owner of ACT-541468. A.A. was an employee of Clinical Research Services Kiel GmbH. There are no other relationships or activities that could appear to have influenced the submitted work. Clinical Research Services Kiel GmbH received financial compensation for the clinical conduct.
Statement of human rights
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Electronic supplementary material
Figure S1
Chemical structure of ACT-541468 (PDF 10 kb)
Figure S2
Arithmetic mean (± SD) (a) trough plasma concentrations of ACT-541468 after multiple-dose administration of ACT-541468 from Day 4 to Day 8, (b) plasma concentration–time profile of ACT-541468 after single- and multiple-dose administration on a linear and semi-logarithmic scale (n = 20) (PDF 170 kb)
Figure S3
Arithmetic mean (± SD) plasma concentration–time profile of (a) midazolam and (b) 1-OH midazolam after administration of midazolam alone, 1 h after single-dose ACT-541468, and after multiple-dose 25 mg ACT-541468 from time 0 to 24 h post-dose on a linear and semi-logarithmic scale (n = 20) (PDF 217 kb)
Rights and permissions
About this article

Cite this article
Boof, ML., Alatrach, A., Ufer, M. et al. Interaction potential of the dual orexin receptor antagonist ACT-541468 with CYP3A4 and food: results from two interaction studies. Eur J Clin Pharmacol 75, 195–205 (2019). https://doi.org/10.1007/s00228-018-2559-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-018-2559-5