
Abstract
Background and Objective
Intracellular exposure of everolimus may be a better marker of therapeutic effect than trough whole blood concentrations. We aimed to develop pharmacokinetic population models and Bayesian estimators based on a limited sampling strategy for estimation of dose interval exposures of everolimus in whole blood and peripheral blood mononuclear cells (PBMCs) in renal transplant recipients.
Methods
Full whole blood and PBMC concentration–time profiles of everolimus were obtained from 12 stable renal transplant recipients on two different occasions, 4 weeks apart. The dataset was treated as 24 individual profiles and split into a development dataset (n = 20) and a validation dataset (n = 4). The pharmacokinetic model was developed using non-parametric modeling and its performances and those of the derived Bayesian estimator were evaluated in the validation set.
Results
A structural two-compartment model with first-order elimination and two absorption phases described by a sum of two gamma distributions were developed. None of the tested covariates (age, sex, albumin, hematocrit, fat-free mass and genetic variants such as CYP3A5*1, ABCB1 haplotype, PPARA*42, PPARA*48, and POR*28) were retained in the final model. A limited sampling schedule of two whole blood samples at 0 and 1.5 h and one PBMC sample at 1.5 h post dose provided accurate estimates of the area under the plasma concentration–time curve (AUC) in comparison with the trapezoidal reference AUC (relative bias ± standard deviation = − 3.9 ± 10.6 and 4.1 ± 12.3% for whole blood and PBMC concentrations, respectively).
Conclusion
The developed model allows simultaneous and accurate prediction of everolimus exposure in whole blood and PBMCs, and supplies a base for a feasible exploration of the relationships between intracellular exposure and therapeutic effects in prospective trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A novel population pharmacokinetic model allowing a joint determination of everolimus in whole blood and peripheral blood mononuclear cells (PBMC) has been developed. |
Bayesian Estimators were developed based on a limited sampling strategy using only two whole blood and one PBMC sample to accurately predict whole blood and intracellular exposure of everolimus. |
The model offers an opportunity to explore the relationship between intracellular everolimus exposure and the clinical effect in prospective trials. |
1 Introduction
The immunosuppressive drug everolimus prevents rejection through inhibition of the mammalian target of rapamycin (mTOR) activity within lymphocytes [1]. Everolimus is characterized by a narrow therapeutic range and a high intra- and inter-individual variability similar to the calcineurin inhibitors (CNIs). Therapeutic drug monitoring (TDM) is therefore recommended for everolimus and, due to its high distribution in erythrocytes, trough whole blood concentrations are usually used for dose individualization [2]. Since the immunosuppressive site of action for everolimus is inside the lymphocyte, it has been hypothesized that the drug concentration in this target compartment would be more directly related to the warranted immunosuppressive effect than whole blood concentrations [3].
The relationship between intracellular and whole blood concentrations of the immunosuppressive drugs has been reported in several publications [4,5,6,7,8,9,10,11]. These studies were performed in small cohorts of patients and usually reported the correlation between whole blood and intracellular trough concentrations without the development of population pharmacokinetic (popPK) models. For the CNIs [cyclosporine (ciclosporin) and tacrolimus], only a weak relationship between intracellular and whole blood concentrations has been reported [4, 5, 7, 12], whereas for the mTOR inhibitors (sirolimus and everolimus), the correlation between the two compartments appears to be stronger (coefficient of determination > 0.56). However, only limited data exist for these drugs [9,10,11]. A few studies have also attempted to investigate the association between intracellular concentrations of CNIs and clinical endpoints such as acute rejection episodes [4, 8]. In these studies, a significantly lower intracellular exposure of cyclosporine and tacrolimus was associated with a higher risk of acute rejection. These results are encouraging and have contributed to the increased interest of monitoring immunosuppressive drugs in their target compartment. However, monitoring intracellular concentrations is time-consuming and technically challenging compared to the standard TDM of whole blood concentrations as it requires isolation in fresh blood and purification of the cells of interest. For practical reasons, peripheral blood mononuclear cells (PBMCs) are generally used as the biological matrix to determine the drug concentration in the target compartment. In addition, intracellular concentration measurement requires an analytical method of sufficient sensitivity and an accurate cell counting system to relate the concentrations obtained to the number of cells. Due to these analytical constraints, the full pharmacokinetic profile measured as area under the plasma concentration–time curve (AUC) to determine the PBMC drug exposure is challenging. However, the a posteriori Bayesian estimation method allows estimation of AUC using popPK models associated with a limited number of samples.
The aim of this study was (1) to develop, using a non-parametric modeling approach, a popPK model allowing a joint determination of whole blood and PBMC concentrations of everolimus; and (2) to build Bayesian estimators (BE) based on a limited sampling strategy (LSS) allowing adequate AUC estimations of everolimus both in whole blood and in PBMC.
2 Materials and Methods
2.1 Patients
Whole blood and PBMC concentrations of everolimus were available from a previous study conducted at the Oslo University Hospital, Rikshospitalet (Oslo, Norway) [13]. Twelve-hour pharmacokinetic profiles from 12 stable renal transplant recipients obtained on two different occasions (4 weeks apart) were included in the present analysis (n = 24 pharmacokinetic profiles). The population, study design, and immunosuppressive protocol have been described in detail previously [9, 13]. In brief, stable renal transplant recipients receiving everolimus, mycophenolate, and corticosteroid-based immunosuppression were included in an open-label, prospective study where the primary aim was to investigate a potential drug–drug interaction between everolimus and the lipid-lowering drug rosuvastatin [13]. Samples for both whole blood and PBMC pharmacokinetic profiles were collected before administration of everolimus (C0) and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, and 12 h after drug intake on both occasions. All patients were genotyped for CYP3A5*3, CYP3A4*22, POR*28, PPARA*42, PPARA*48, and ABCB1 variants (1236C>T, 2677G>T, 3435C<T), as previously described [9, 13]. The study was performed in accordance with the Declaration of Helsinki, and written informed consent was obtained from all the patients before inclusion. The study was approved by the Regional Committee for Medical and Health Research Ethics and by the Norwegian Medicines Agency (EudraCT number: 2011-005212-29). The study is registered in ClinicalTrials.gov (NCT01524601).
2.2 Isolation of Peripheral Blood Mononuclear Cells (PBMCs) and Drug Analysis
PBMCs were isolated from freshly collected whole blood using LeucoSep™ tubes (Greiner Bio-One, Frickenhausen, Germany) as previously described [9]. Samples were drawn in pre-chilled tubes, placed immediately on ice, and the whole isolation procedure was performed at 4 °C to avoid passive diffusion of everolimus from the PBMCs. Cell counting was performed (10 ml) on a Beckman Coulter Counter® (Beckman Coulter, Brea, CA) applying a diameter range from 5 to 15 mm. An accurate volume of the cell suspension was transferred to a microcentrifuge tube. After centrifugation, the supernatant was removed and the pellet was stored at − 70 °C until liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis. The concentrations of everolimus in PBMC were normalized to 106 cells. Both whole blood and PBMC concentration of everolimus were determined with a validated LC–MS/MS assay, as previously described [9, 14].
2.3 Pharmacokinetic Modeling
Modeling was performed using the non-parametric adaptive grid approach implemented in R software (Pmetrics version 1.5.0) [15]. Data were randomly split into a development dataset (n = 20 profiles) and a validation dataset (n = 4 profiles). Splitting into respective group was based on a simple randomization table, assuming all 24 pharmacokinetic profiles to be independent from one another. In the initial model development phase, different models to describe the absorption phase of everolimus were investigated and compared, including a two-compartment model with first-order absorption and lag time as well as gamma distributions.
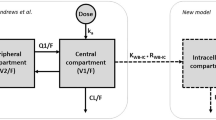
A two-compartment model with whole blood and PBMC as the first and second compartment, respectively, and a double gamma distribution to describe the absorption into compartment one was investigated [16, 17]:
where f(t) is the absorption rate, F is the bioavailability factor, D is the administered dose, r is the fraction of dose which transits via the first absorption route, and f1 and f2 are the probability density functions of the two gamma distributions, such as:
where ai and bi are the parameters of the distributions (i = 1 or 2) and Γ is the classical gamma function.
The drug amounts in the two compartments are given by Eq. 12 in the Electronic Supplementary Material, as follows:
where x(t) is the vector of drug amounts in the two compartments, α and β are the apparent rate constants for the distribution and elimination phases, k12 and k21 are the transfer rate constants between the two compartments; (Ik × f)(t) denotes the convolution product of the exponential function Ik(t) = exp(–kt) with the rate function f(t), where k = α or β.
The theoretical concentrations C1(t) and C2(t) in the two compartments are computed by dividing the amount in the respective compartment by the corresponding volume of distribution.
The measured whole blood concentration C1obs(t) was directly compared with the theoretical concentration C1(t), and the measured PBMC concentration C2obs(t) was set to be proportional to the theoretical concentration C2(t):
where q is a proportionality factor, which is necessary because the two-compartment model defines the volume of distribution V2 so that the mean steady-state concentrations in the two compartments are equal, which of course is not necessarily the case with the observed concentrations. This model is described in detail in the Electronic Supplementary Material.
For the error model, in order to weight the concentrations by the reciprocal of their variances in the fitting process we used a polynomial error of the following form for both the whole blood concentrations and PBMC concentrations:
where SD is the standard deviation of the concentration and conc is the measured everolimus concentration.
Additionally, we allowed Pmetrics to fit an additive term λ, so that concentrations were weighted as follows:
where λ is representative of additional noise.
Model selection was based on comparison of the Akaike information criterion (AIC) and minimization of the bias and imprecision of the individual predictions versus observations. Associations between individual pharmacokinetic parameters and different covariates were examined by multiple linear regression and graphical examination in the development dataset. Total body weight, age, sex, albumin, hematocrit, fat-free mass, as well as different genetic variants that may influence everolimus pharmacokinetics including CYP3A5*1, CYP3A4*22, ABCB1 haplotype (1236C>T, 2677G>T, 3535C>T), PPARA*42, PPARA*48, and POR*28 were investigated as covariates. Since only one of the patients had the CYP3A4*1/*22 variant, this genotype was not investigated in the current study. If a significant association was observed (p < 0.01) between the pharmacokinetic parameter and covariates, they were introduced in the model. The covariates were normalized to the population median values, with continuous covariates modelled using the following general equation:
where θi is the individual model-predicted pharmacokinetic parameter for an individual with covariate value covi, θpop represents the population central tendency for the pharmacokinetic parameter θ, covm represents the population median value of the covariate, and θcov represents the covariate effect. The importance of each covariate was evaluated by comparing AIC, imprecision, and bias of the model with or without the covariate of interest.
Diagnostic plots were drawn for the final model with individual predictions versus observed concentrations and weighted residues versus observed concentrations. Since different dosages were used in the study, the internal evaluation of the final model was performed using the prediction-corrected visual predictive checks (pcVPCs). The pcVPCs were based on 1000 Monte-Carlo simulations. In a pcVPC, the variability coming from binning across independent variables is removed by normalizing the observed and simulated dependent variable based on the typical population prediction for the median independent variable in the bin [18]. The observed data were overlaid on the prediction intervals and compared visually.
2.4 Development of Bayesian Estimators
The final model obtained from the development dataset was used to determine the best LSS using the multiple model optimal sampling function (MMopt) weighted for AUC included in Pmetrics. This sampling algorithm chooses the sample times which minimize the risk of misrepresenting the patient as the wrong set of support points in the model, i.e. estimating the wrong set of pharmacokinetic parameters for the patient [19]. We investigated different numbers of optimal samples (n = 1, 2, or 3) for both whole blood and PBMC, restricted to a sampling time between zero and 4 h after dose administration for practical reasons. Our goal was to minimize the number of PBMC samples while still ensuring accurate estimates of the patients’ whole blood and PBMC AUC of everolimus.
The predictive performance of the BEs developed using different LSS was evaluated in the validation dataset by comparing the inter-dose AUC from time zero to 12 h (AUC12) obtained using the LSS to the reference AUC12 obtained using the linear trapezoidal method applied to the full pharmacokinetic profiles. A calculation of the root mean square error (RMSE), relative bias, and number of profiles from the ± 20% interval compared with the reference AUC12 was performed. The LSS with a combination of the lowest relative bias and RMSE (< 15%) and zero patients outside of the ± 20% interval in both whole blood and PBMC was considered to be best.
3 Results
3.1 Structural Model Development
The characteristics of the patient population used for the model are shown in Table 1.
A structural two-compartment model with whole blood and PBMC everolimus concentrations as the first and second compartment, respectively, two absorption phases described by a sum of two gamma distributions into the whole blood compartment, and first-order elimination from the whole blood compartment described the data well. The gamma distribution model was superior to the other absorption model (first-order absorption with lag time) tested in the initial development phase both in terms of AIC (1604 versus 1081 for the first-order absorption with lag time model and gamma distribution model, respectively) and individual predictions (data not shown). The final model had 19 support points where each support point is a vector of estimates for each parameter value, and of the probability of the combination of these values. There can be at most one support point for each subject in the study population. The population pharmacokinetic parameters are presented in Table 2. The median (range) whole blood AUC12 was 72.0 (50.4–134.8) µg h l−1 and median apparent clearance estimated as AUC12/dose was 14.7 (7.4–39.7) l h−1. The two best and the two worst fits of individual pharmacokinetic profiles in the development dataset using all time points for whole blood and PBMC concentration prediction of everolimus are presented in Fig. 1. Individual predicted pharmacokinetic profiles for all patients in the development dataset are presented in Electronic Supplementary Material Fig. 1.

The two best (a, b) and two worst (c, d) individual predicted profiles in the development dataset for everolimus concentrations in whole blood and in peripheral blood mononuclear cells (PBMCs). The black lines are the model predicted curves using all timepoints. The light grey crosses are the observed PBMC concentrations and the black crosses are the observed whole blood concentrations of everolimus
3.2 Covariate Investigation and Internal Validation
Total body weight was significantly associated with the scale of the first gamma law, b1 (p = 0.00995) corresponding to a faster absorption [as mean absorption time (MAT) = a/b, if b increases, MAT decreases]. However, its introduction in the model worsened the AIC and did not improve the model predictions. None of the other covariates tested, including the selected polymorphism in genes coding for enzymes/transporters involved in distribution and metabolism of everolimus, were significantly associated with any pharmacokinetic parameters. The individual predicted versus observed plots for whole blood and PBMC concentrations are shown in Fig. 2. The regression coefficient of determination (R2) was 0.97 and 0.89 and the slopes were 1.01 [95% confidence interval (CI) 0.98–1.04] and 1.08 (95% CI 1.03–1.13) for the whole blood and PBMC concentrations, respectively. There was no major bias and the weighted residuals were homogeneously distributed over the concentration range for both whole blood and PBMC concentrations (Fig. 2).

Diagnostic plots: individual predicted whole blood and peripheral blood mononuclear cell (PBMC) concentrations as a function of observed whole blood (a) and PBMC concentrations (b), population predicted whole blood and PBMC concentrations as a function of observed whole blood (c) and PBMC concentrations (d), and weighted residuals as a function observed whole blood (e) and PBMC concentrations (f)
The relative bias ± SD (RMSE) between observed concentrations and concentrations estimated using the final model was: − 0.19 ± 9.62% (9.60%) for the whole blood concentrations and − 0.14 ± 16.5% (16.5%) for the PBMC concentrations. The performance of the model was evaluated with a pcVPC is shown in Fig. 3. The normalized observed data adequately overlaid the 90% prediction intervals of the simulations for both whole blood and PBMC concentrations.

Predicted corrected visual predictive checks for whole blood (a) and peripheral blood mononuclear cell (PBMC) (b) concentrations of everolimus in renal transplant recipients
3.3 Relationship Between Everolimus Concentrations in Whole Blood and PBMCs
The model predicted, after an appropriate amount of time, that the concentrations C1 and C2 in the two compartments became proportional. This is exemplified in Fig. 4, which shows the time course of the two concentrations and the relationship between them. The relationship between the two concentrations became linear after about 24 h when C1 < 0.5 µg l−1.

Time course of everolimus concentrations (C) in the two compartments (a solid line: whole blood concentration (C1), dashed line: peripheral blood mononuclear cell concentration [C2]) and the relationship between them (b time progression is indicated by arrows). C1is concentrations in compartment 1, C2is concentrations in compartment 2
3.4 Limited Sampling Strategy
The popPK parameters obtained from the development dataset were used as priors for the development of the BE in the validation dataset. Table 3 (and Electronic Supplementary Material Fig. 2) shows the results of the different types of LSS tested (with different numbers and combinations of samples), based on the results of the MMopt algorithm. The BE derived from the final model and based on whole blood and PBMC samples obtained at 0, 1.5, and 3 h after dose administration (LSS 9) provided the best combination of estimates for both the whole blood and PBMC everolimus AUC12 in the validation dataset. Using only whole blood samples (0, 1, and 3 h), the estimated PBMC everolimus AUC12 (LSS 2) was worse with a RSME of 26.7% and two of four patients fell outside the ± 20% range. Finally, BE based on an LSS of two whole blood samples at 0 (C0) and 1.5 h (C1.5) and one PBMC sample at 1.5 h (C1.5) (LSS12) provided an estimation of AUC12 with a relative bias ± SD of − 3.9 ± 10.6 and 4.1 ± 12.3% for whole blood and PBMC everolimus AUC12, respectively (Table 3, Electronic Supplementary Material Fig. 2). This LSS was considered to be satisfactory due to the low number of both whole blood and PBMC samples needed to provide estimates with RSME < 15% and zero patients outside the ± 20% interval in both matrices.
4 Discussion
In the current study we developed (1) a popPK model for jointly modeling the whole blood and PBMC concentrations of everolimus; and (2) a BE based on an LSS using only two whole blood (C0 and C1.5) and one PBMC (C1.5) sample to accurately predict everolimus whole blood and PBMC exposure in renal transplant recipients.
The median everolimus apparent clearance estimated from our model (14.7 l h−1) was consistent with the value reported in a previously published popPK model of everolimus in renal transplant recipients (17.9 l h−1) [20]. The estimated mean AUC was approximately twofold higher than the AUC estimates from our study, but this is likely due to the different target ranges used for dose individualization between the two studies [20]. A model in heart transplant recipients reported a lower apparent clearance of everolimus (3.33 l h−1) [21]. However, co-administration of cyclosporine, which is known to influence everolimus pharmacokinetics, a different patient population, and differences in modeling in this study are probably the reasons for the observed discrepancies.
To the best of our knowledge, no popPK model with an associated LSS to predict AUC values both in whole blood and at the target site (i.e. within PBMCs) has been reported to date for any of the immunosuppressive drugs used after solid organ transplantation. The ability to model PBMC concentrations based on sparse blood sampling offers an opportunity to explore the relationship between PBMC exposure and the clinical effect in prospective trials, and is also highly relevant for all immunosuppressive drugs. In the model developed from the present study, a linear relationship is drawn between the inter-dose AUC in whole blood and in PBMCs at steady state. In most of the previous published studies, simple linear relationships between whole blood and PBMC trough concentrations of the immunosuppressive drugs are reported and exhibit weak correlations [3]. However, in our model, a linear relationship between drug concentrations in the two compartments is applicable, but only after the first dose and during the elimination phase when the influence of the alpha exponential becomes negligible.
Developing an LSS based on a combination of a total three samples limited to the first 4 h post-dose seems to be a good compromise between the precision of the parameter estimates and a possible implementation in routine practice or clinical trials. It was, as expected, not possible to use our model to estimate the PBMC exposure accurately using only the whole blood concentrations. This indicates that intracellular monitoring is worth investigating as a potential alternative to standard whole blood monitoring of everolimus. The timepoints in our proposed LSS is in agreement with previous studies conducted in heart or renal transplant recipients, which showed an accurate estimation of everolimus inter-dose AUC values using only two blood samples (C0 and C2) [20, 22].
Although TDM has contributed to optimize clinical outcomes, transplanted patients still experience acute rejection episodes and toxicity despite trough whole blood drug concentrations in the target range [23]. Monitoring drug concentrations at the target site, i.e. in lymphocytes, has therefore been investigated as an alternative method to monitor the effect and toxicity of the immunosuppressive drugs [3]. Most of the studies published regarding this topic have focused on the relationship between whole blood and intracellular drug trough concentrations [5,6,7, 9, 10]. As the everolimus concentrations in whole blood and PBMC have previously been shown to be associated, the usefulness of intracellular monitoring for this drug has been questioned [24]. However, only limited data from small cohorts are available and it is still unclear if intracellular concentrations could serve as better surrogate markers than current blood TDM [24]. The potential interest of intracellular concentrations would significantly increase if a relationship with a clinical endpoint, such as acute rejection or drug toxicity, could be established [4, 8], and the clinical significance of intracellular immunosuppressive drugs measurement therefore needs to be evaluated in large prospective trials. Due to the analytical constraints associated with intracellular measurements, it has been a challenge to implement it in a routine setting or large clinical trials. This can now be overcome by using the LSS that we have developed in our study which may make it easier to investigate the relationships between PBMC everolimus exposure and clinical outcomes (e.g., rejections).
Further studies should investigate the influence of everolimus concentrations in PBMC on pharmacodynamics targets in the mTOR pathway (i.e. p70 ribosomal S6 kinase [S6K1]). Indeed, as S6K1 inhibition is considered to be a good biomarker for monitoring mTOR inhibition [25], investigations regarding the effect of everolimus concentrations in both PBMC and whole blood on measured p70S6K inhibition or risk of acute rejection/toxicity could provide relevant results in relation to clinical endpoints. However, this was not foreseen when we designed our study and we unfortunately did not collect any relevant samples to perform p70S6K inhibition measurements. Model-based simulation can be utilized to explore the probability of attaining a pharmacokinetic/pharmacodynamic target and may be useful to increase the knowledge of the effect/exposure relationship of everolimus. In fact, we did initially plan to perform model-based simulations to explore the probability of attaining a pharmacokinetic/pharmacodynamic target based on the inhibition by everolimus of the p70S6 kinase. However, after an extensive search of the literature we were not able to find relevant everolimus IC50 (the half maximal inhibitory concentration) value for this target in renal transplant patients (or other solid organ transplantation). Most of the publications describe the p70S6K inhibition by everolimus in cancer patients or cancer cell lines, and in these studies the treatment regimens and everolimus concentrations were quite different from those used in transplanted patients [25,26,27].
In agreement with previously published studies using the gamma distribution model to describe the absorption process of immunosuppressive drugs, no covariates were retained in the final model [16, 28, 29]. This is in contrast to previous popPK models of everolimus described in the literature where covariates such as ideal body weight and bilirubinemia have been shown to influence everolimus pharmacokinetics [20, 21]. Consistent with the previous observations, polymorphisms in relevant genes coding for CYP3A5 and ABCB1 did not have an effect on everolimus pharmacokinetics in the present study [20]. However, Picard et al. [30] showed in vitro that cytochrome P450 (CYP) 3A4 was a better catalyst of everolimus metabolism than CYP3A5. Since only one of the patients had CYP3A4*1/*22, this genotype was not investigated in the current study.
Since 75% of everolimus is distributed into erythrocytes, red blood cell contamination in isolated PBMC samples can influence the concentration measurements [2]. In one of the patients included, most of the PBMC samples at one of the pharmacokinetic profile occasions showed a high degree of hemolysis. This may have contributed to the poorly predicted individual PBMC profile shown in Fig. 1c, as the concentrations of everolimus may have been falsely high in these PBMC samples.
The strength of our study is the number of rich pharmacokinetic profiles collected both for whole blood and PBMC everolimus concentrations. Indeed, it is particularly difficult to obtain a large number of PBMC samples as it requires laborious and time-consuming sample handling. The structural model developed in the present study may be generalized to apply for the other immunosuppressive drugs as well. This would obviously imply that a sufficient dataset describing the pharmacokinetics of the relevant drug in both matrices is available. Our study also has some limitations. First, only four pharmacokinetic profiles were used to validate the predictive performance of the developed model. Unfortunately, no other external dataset with everolimus concentrations in PMBC was available, and further investigations should include validation using data from a larger cohort of patients with everolimus concentrations both in whole blood and PBMC. Secondly, the patients’ pharmacokinetic profiles for everolimus were obtained before and after 1 month of treatment with rosuvastatin. However, the treatment with rosuvastatin did not affect either the whole blood or the PBMC pharmacokinetics of everolimus [13].
5 Conclusion
We have successfully developed a popPK model and BE based on an LSS for whole blood and PBMC concentrations of everolimus. This model allows a simultaneous and accurate determination of whole blood and PBMC exposure of everolimus, and may also provide a good basis for development of similar models for other immunosuppressive drugs. More knowledge regarding the potential impact of monitoring concentrations of the immunosuppressive drugs at their target sites is required and by using our model, the relationship between PBMC exposure and therapeutic effect of these drugs may be easier to determine.
References
Schuler W, Sedrani R, Cottens S, Häberlin B, Schulz M, Schuurman HJ, et al. SDZ RAD, a new rapamycin derivative: pharmacological properties in vitro and in vivo. Transplantation. 1997;64(1):36–42.
Kirchner G, Meier-Wiedenbach I, Manns M. Clinical pharmacokinetics of everolimus. Clin Pharmacokinet. 2004;43(2):83–95.
Lemaitre F, Antignac M, Verdier MC, Bellissant E, Fernandez C. Opportunity to monitor immunosuppressive drugs in peripheral blood mononuclear cells: where are we and where are we going? Pharmacol Res. 2013;74:109–12.
Falck P, Asberg A, Guldseth H, Bremer S, Akhlaghi F, Reubsaet JL, et al. Declining intracellular T-lymphocyte concentration of cyclosporine a precedes acute rejection in kidney transplant recipients. Transplantation. 2008;85:179–84.
Lemaitre F, Antignac M, Fernandez C. Monitoring of tacrolimus concentrations in peripheral blood mononuclear cells: application to cardiac transplant recipients. Clin Biochem. 2013;46(15):1538–41.
Lepage JM, Lelong-Boulouard V, Lecouf A, Debruyne D, Hurault de Ligny B, Coquerel A. Cyclosporine monitoring in peripheral blood mononuclear cells: feasibility and interest. A prospective study on 20 renal transplant recipients. Transplant Proc. 2007;39(10):3109–10.
Lemaitre F, Blanchet B, Latournerie M, Antignac M, Houssel-Debry P, Verdier MC, et al. Pharmacokinetics and pharmacodynamics of tacrolimus in liver transplant recipients: inside the white blood cells. Clin Biochem. 2015;48(6):406–11.
Capron A, Lerut J, Latinne D, Rahier J, Haufroid V, Wallemacq P. Correlation of tacrolimus levels in peripheral blood mononuclear cells with histological staging of rejection after liver transplantation: preliminary results of a prospective study. Transpl Int. 2012;25:41–7.
Robertsen I, Vethe NT, Midtvedt K, Falck P, Christensen H, Åsberg A. Closer to the site of action: everolimus concentrations in peripheral blood mononuclear cells correlate well with whole blood concentrations. Ther Drug Monit. 2015;37(5):675–80.
Roullet-Renoleau F, Lemaitre F, Antignac M, Zahr N, Farinotti R, Fernandez C. Everolimus quantification in peripheral blood mononuclear cells using ultra high performance liquid chromatography tandem mass spectrometry. J Pharm Biomed Anal. 2012;66:278–81.
Masri M, Rizk S, Barbari A, Stephan A, Kamel G, Rost M. An assay for the determination of sirolimus levels in the lymphocyte of transplant patients. Transplant Proc. 2007;39(4):1204–6.
Robertsen I, Falck P, Andreassen AK, Næss NK, Lunder N, Christensen H, et al. Endomyocardial, intralymphocyte, and whole blood concentrations of ciclosporin A in heart transplant recipients. Transplant Res. 2013;2(1):5.
Robertsen I, Asberg A, Granseth T, Vethe NT, Akhlaghi F, Ghareeb M, et al. More potent lipid-lowering effect by rosuvastatin compared with fluvastatin in everolimus-treated renal transplant recipients. Transplantation. 2014;97(12):1266–71.
Vethe NT, Gjerdalen LC, Bergan S. Determination of cyclosporine, tacrolimus, sirolimus and everolimus by liquid chromatography coupled to electrospray ionization and tandem mass spectrometry: assessment of matrix effects and assay performance. Scand J Clin Lab Invest. 2010;70(8):583–91.
Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit. 2012;34(4):467–76.
Debord J, Risco E, Harel M, Le Meur Y, Büchler M, Lachâtre G, et al. Application of a Gamma model of absorption to oral cyclosporin. Clin Pharmacokinet. 2001;40(5):375–82.
Nagy G. Ordinary differential equations. 2017. http://users.math.msu.edu/users/gnagy/teaching/ode.pdf. Accessed 26 Oct 2017
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51.
Bayard DS, Neely M. Experiment design for nonparametric models based on minimizing Bayes Risk: application to voriconazole. J Pharmacokinet Pharmacodyn. 2017;44(2):95–111.
Moes DJ, Press RR, den Hartigh J, van der Straaten T, de Fijter JW, Guchelaar HJ. Population pharmacokinetics and pharmacogenetics of everolimus in renal transplant patients. Clin Pharmacokinet. 2012;51(7):467–80.
Lemaitre F, Bezian E, Goldwirt L, Fernandez C, Farinotti R, Varnous S, et al. Population pharmacokinetics of everolimus in cardiac recipients: comedications, ABCB1, and CYP3A5 polymorphisms. Ther Drug Monit. 2012;34(6):686–94.
Terada Y, Wada K, Matsuda S, Kuwahara T, Kawabata A, Takada M, et al. Circadian pharmacokinetics and limited sampling strategy of everolimus in heart transplant patients. Int J Clin Pharmacol Ther. 2017;55(1):1–8.
Oellerich M, Armstrong VW. The role of therapeutic drug monitoring in individualizing immunosuppressive drug therapy: recent developments. Ther Drug Monit. 2006;28(6):720–5.
Capron A, Haufroid V, Wallemacq P. Intra-cellular immunosuppressive drugs monitoring: a step forward towards better therapeutic efficacy after organ transplantation? Pharmacol Res. 2016;111:610–8.
O’Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOR inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol. 2010;3(2):65–79.
Leung EY, Askarian-Amiri M, Finlay GJ, Rewcastle GW, Baguley BC. Potentiation of growth inhibitory responses of the mTOR inhibitor everolimus by dual mTORC1/2 inhibitors in cultured breast cancer cell lines. PLoS One. 2015;10(7):e0131400.
Tanaka C, O’Reilly T, Kovarik JM, Shand N, Hazell K, Judson I, et al. Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol. 2008;26(10):1596–602.
Woillard JB, Debord J, Monchaud C, Saint-Marcoux F, Marquet P. Population pharmacokinetics and Bayesian estimators for refined dose adjustment of a new tacrolimus formulation in kidney and liver transplant patients. Clin Pharmacokinet. 2017;56(12):1491–8.
Saint-Marcoux F, Knoop C, Debord J, Thiry P, Rousseau A, Estenne M, et al. Pharmacokinetic study of tacrolimus in cystic fibrosis and non-cystic fibrosis lung transplant patients and design of Bayesian estimators using limited sampling strategies. Clin Pharmacokinet. 2005;44(12):1317–28.
Picard N, Rouguieg-Malki K, Kamar N, Rostaing L, Marquet P. CYP3A5 genotype does not influence everolimus in vitro metabolism and clinical pharmacokinetics in renal transplant recipients. Transplantation. 2011;91(6):652–6.
Acknowledgements
The authors thank Mrs Karen Poole of the Department of Pharmacology, Toxicology and Pharmacovigilance, CHU Limoges for manuscript editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors did not receive any funding for this project.
Conflict of interest
I. Robertsen, J. Debord, A. Åsberg, K. Midtvedt, P. Marquet, and J.-B. Woillard declare that they have no conflicts of interest.
Research involving human participants
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Robertsen, I., Debord, J., Åsberg, A. et al. A Limited Sampling Strategy to Estimate Exposure of Everolimus in Whole Blood and Peripheral Blood Mononuclear Cells in Renal Transplant Recipients Using Population Pharmacokinetic Modeling and Bayesian Estimators. Clin Pharmacokinet 57, 1459–1469 (2018). https://doi.org/10.1007/s40262-018-0646-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-0646-5