
Abstract
Breast cancer is a heterogeneous disease, manifesting in a broad differentiation in phenotypes and morphologic profiles, resulting in variable clinical behavior. Between 10 and 20% of all breast cancers are triple negative. Triple-negative breast cancer (TNBC) lacks the expression of human epidermal growth factor receptor 2 (HER2) and hormone receptors; therefore, to date, chemotherapy remains the backbone of treatment. TNBC tends to be aggressive and has a high histological grade, resulting in a poor 5-year prognosis. It has a high prevalence of BRCA1 mutations and an increased Ki-67 expression. This subtype usually responds well to taxanes and/or platinum compounds and poly (ADP-ribose) polymerase (PARP) inhibitors. Studies with PARP inhibitors have demonstrated promising results in the treatment of BRCA-mutated breast and ovarian cancer, and PARP inhibitors have been studied as monotherapy and in combination with cytotoxic therapy or radiotherapy. PARP inhibitor efficacy on poly (ADP-ribose) polymer (PAR) formation in vivo can be quantified by pharmacodynamic assays that measure PAR activity in peripheral blood mononuclear cells (PBMC). Biomarkers such as TP53, ATM, PALB2 and RAD51C might be prognostic or predictive indicators for treatment response, and could also provide targets for novel treatment strategies. In summary, this review provides an overview of the treatment options for basal-like TNBC, including PARP inhibitors, and focuses on the pharmacotherapeutic options in these patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Triple negative breast cancer is a subgroup of breast cancer with poor prognosis for which there are only few treatment options of proven benefit, necessitating exploration of novel targets for drug therapy. |
PARP inhibitors are a group of novel oral anticancer drugs highly active in a subgroup of triple negative breast cancer with selected mutations or epigenetic silencing of genes involved in the DNA damage response (DDR), including BRCA1 and BRCA2. |
Pharmacotherapy in the case of triple negative breast cancer is involved with developing novel therapies focusing on, amongst others, PARP-inhibitors. |
1 Introduction
Breast cancer is the most common cancer in women worldwide [1], and, unfortunately, the incidence of breast cancer is still rising worldwide. In the past few decades, enormous progress has been made in the understanding of the molecular pathways involving breast cancer, leading to the development of more personalized therapies; however, despite this, the 5-year survival of metastatic breast cancer remains low [2]. Breast cancer is a very heterogeneous disease, manifesting in a broad differentiation in phenotypes and morphologic profiles, resulting in different clinical behaviors [3]. Based on their immunohistochemical features, breast cancers can be divided into three main types: hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-positive, and triple-negative (TN) tumors [4]. Between 10 and 20% of breast cancers are triple negative. TN breast cancers (TNBCs) are characterized by the absence of estrogen receptor (ER), progesterone receptor (PR), and HER2 expression [5]. TNBC tends to be aggressive, occurs at a younger age, and has a higher grade. It has a high recurrence rate and a poor 5-year prognosis compared with other types of breast cancer [6]. TNBCs cannot be treated with targeted therapy such as endocrine therapy or trastuzumab because they lack cellular targets [7]; therefore, treatment of TNBC remains challenging. In this review, we describe the molecular features of TNBC, the relationship with BRCA mutation status, and treatment with a novel class of anticancer agents, the poly (ADP-ribose) polymerase (PARP) inhibitors. We also discuss patient selection, biomarkers, and individualization of dosing schedules employing new pharmacodynamic assays.
2 Homologous Recombination Deficiency
If the process of homologous recombination (HR) is unavailable or impaired, this is referred to as ‘homologous recombination deficiency’ (HRD). In this situation, DNA repair is more error prone, which leads to genomic instability [8, 9]. Bunting et al. showed that loss of 53BP1 restored HR activity in BRCA1-mutant cells with HR deficiency [10]. In addition, using a cell-based screen, Bouwman et al. showed that 53BP1 is essential for continuing the growth arrest induced by BRCA1 mutation [11]. Other mechanisms of resistance to BRCA-targeted therapies could be through secondary mutations, which could restore BRCA1 and BRCA2 function. These secondary somatic mutations predict resistance to platinum chemotherapy and PARP inhibitors in women with BRCA1/2 mutations [12, 13]. Tumors with HRD can be sensitive to DNA cross-linking agents, such as alkylators and platinum drugs [14,15,16]. BRCA1 and BRCA2 play a pivotal role in the repair of double-strand breaks (DSBs) in DNA by the process of HR. Loss of either BRCA1 or BRCA2 function leads to HRD [17]; however, sensitivity to alkylators and other agents can be lost if 53BP1 is lost in addition to impaired BRCA function. At present it is unclear whether the presence of HRD is a requirement for tumor cells to be sensitive to alkylating agents, platinum compounds, and other drugs. In addition, it is unclear which diagnostic test best enriches tumors with HRD. Finally, it is unclear what is most needed in the clinic—a test that reliably measures HRD or a test that reliably measures sensitivity to a certain agent, or a combination of agents.
3 BRCA Mutations and Other Potential Homologous Recombination Deficiency-Inducing Mutations
Approximately 10% of newly diagnosed TNBC patients harbor a mutation in genes encoding for breast cancer susceptibility protein 1/2 (BRCA1/2) [18,19,20]. A large portion of TN breast neoplasms have similar characteristics as tumors that harbor a germline BRCA1 or BRCA2 mutation, and more than 80% of breast cancer patients with a germline BRCA1 mutation have a TN breast tumor [21].
Tumors that harbor a BRCA mutation are often highly sensitive to drugs that induce DNA DSBs, such as alkylating agents, and less sensitive to spindle poisons [22]. Treating TNBC patients with platinum compounds is based on the fact that TNBC has molecular similarities to BRCA-mutated breast cancers, which are sensitive to platinum compounds [23]. In addition to sensitivity to platinum compounds, TN tumors and BRCA1-mutant breast cancers have various concordances, such as a basal-like profile, frequent TP53 mutations, and a high load of genomic aberrations such as loss of heterozygosity [24].
In a phase II trial, Byrski et al. showed that cisplatin chemotherapy is highly active in women with a BRCA1 germline mutation [25], and that platinum-based chemotherapy is effective in a high proportion of patients with BRCA1-associated cancer [26]. Tumors without germline BRCA1 mutation that have absent or reduced BRCA1 expression may be linked to hypermethylation of the BRCA1 promotor region [27]. Approximately 9.1–37% of sporadic breast cancers have hypermethylation of BRCA1, a condition that is associated with high tumor grade, ER negativity, basal marker expression, younger age at diagnosis, and reduction or loss in BRCA1 messenger RNA (mRNA) expression [8, 27,28,29]. In the absence of a BRCA1 mutation, BRCA1 promotor hypermethylation could be an indicator of an impaired BRCA function [30].
Two neoadjuvant clinical trials have shown that part of the sporadic, non-BRCA1-mutated TNBC is sensitive to platinum compounds [24]. Heterogeneity of TNBC makes treatment challenging, and gathering more insight into heterogeneity could lead to improved and more focused therapy [31]. Rare inactivating mutations in several genes in the DSB repair pathway are associated with the development of cancer. These genes, like RAD51c, ATM, and PALB2, are involved in a small fraction of the disease. RAD51c works with BRCA1 and BRCA2 to repair DNA DSBs, and the overall mutation frequency of RAD51c in familial breast cancer is low [32]. PALB2 is a breast cancer susceptibility gene that interacts with BRCA2, while ATM interacts with BRCA1 [33]. Depletion of ATM in breast cancer cells could sensitize these cells to PARP inhibition, which suggests a treatment potential for breast cancers with low ATM protein expression [34].

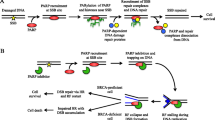
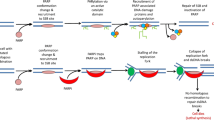
PARP inhibition. PARP poly (ADP ribose) polymerase. From Sonnenblick et al. [82]
4 Molecular Features of Triple-Negative Breast Cancer
Recently, six subtypes of TNBC have been identified: basal-like 1 (BL-1), basal-like 2 (BL-2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like (MSL), and luminal androgen receptor (LAR)-positive [6]. Understanding of and insight into these molecular subtypes could contribute to better treatment strategies due to more individualized treatments. The BL-1 subtype is characterized by an enriched cell cycle and DNA damage response gene expression [35].
Enrichment in proliferation genes and increased Ki-67 expression in basal-like TNBC could explain why this subtype responded well to antimitotic agents such as taxanes [36, 37], although the notion that high proliferative tumors respond better to spindle poisons contradicts with findings in the Oxford Overview, where patients with well-differentiated ER-positive breast cancers appeared to benefit more from taxane-containing regimens than patients with moderately and poorly differentiated ER-positive breast cancers in the adjuvant setting [38]. Either response rate does not match with long-term survival or spindle poisons do not only frustrate mitosis but also have another, yet unknown, mechanism of action.
Two clinical trials showed that patients who received taxanes as neoadjuvant therapy had a higher pathological complete response (pCR) rate (63%; p = 0.042) when the basal-like subtype was compared with the mesenchymal-like type (31%) or LAR (14%) type [6]. Masuda et al. showed that BL-1 tumors have the highest rate of pCR (52%) with taxane-based neoadjuvant regimens, compared with other subtypes [39]. Basal-like breast cancers show a high prevalence of BRCA1 mutations [40]. In their study, Turner et al. found that 63% of metaplastic breast cancers, a rare type of basal-like cancers, had BRCA1 promoter methylation, compared with 12% in the control group (p < 0.0001). This high incidence of BRCA1 methylation might point to a new treatment strategy for patients with basal-like breast cancer [41].
Basal-like TNBC has similarities to BRCA1-mutated tumors, such as morphological features and immunohistochemical profile, like a similar pattern of cell cycle protein expression [42, 43]. Therefore, another promising target for the treatment of BL-1 breast tumors are PARP inhibitors. Sensitivity for particular agents is not restricted to BRCA-mutated tumors. It is thought that approximately 30% of sporadic breast cancers also have defects in HR repair, a phenotype that is referred to as ‘BRCAness’. Several studies have shown that breast cancer that harbors a BRCA1 or BRCA2 mutation has a characteristic pattern of DNA gains and losses in an array comparative genomic hybridization (aCGH) assay [44,45,46,47]. Vollebergh et al. showed that a subgroup of HER2-negative tumors characterized by a BRCA1-like aCGH (BRCA1-likeCGH) pattern had benefit from high-dose platinum therapy. Patients with BRCA1 loss (not BRCA-mutated) can be found by aCGH, and this could thereby identify patients who could have benefit from DNA DSB-inducing chemotherapy [48]. Lips et al. identified a BRCA2-like CGH (BRCA2-likeCGH) pattern and found this to be present in some sporadic breast cancers [8]. The BRCA2-likeCGH pattern was in contrast to the BRCA1-likeCGH pattern frequently observed in ER-positive tumors.
In a follow-up study, Vollebergh et al. explored not only ER-negative but also ER-positive breast cancer patients who could be identified as benefiting from DNA cross-linking agents. Fifty-one percent (41/81) of the BRCA-likeCGH tumors were ER-positive, showing that patients with BRCA2-likeCGH tumors have more benefit from intensified DNA DSB-inducing agents when compared with standard chemotherapy, like patients with the BRCA1-likeCGH pattern [49]. The BRCA-likeCGH could be helpful in selecting patients who may have benefit from intensified DNA DSB-inducing agents in combination with autologous stem cell rescue.
5 PARP Inhibitors
PARP is a damage recognition repair protein of a single-strand break (SSB) and plays an important role in initiation of the repair of SSBs in the DNA by base excision repair. Inhibition of PARP results in accumulation of SSBs, which can lead to the formation of DSBs. Cells that are BRCA-mutant are not able to repair DSBs error free, which can ultimately lead to cell death. When a deficiency in one gene does not lead to cell death, but a combination of two or more deficiencies do, such as the combination of a PARP inhibitor and a BRCA mutation, this is called synthetic lethality [50] (Fig. 1).
5.1 Olaparib
Olaparib is a potent oral PARP inhibitor that can be lethal to cells harboring a BRCA1 or BRCA2 mutation [51]. In a phase I trial, Fong et al. demonstrated the pharmacokinetic and pharmacodynamic characteristics of the olaparib capsule formulation, with pharmacokinetic parameters of twice-daily dosing showing fast absorption and elimination. The peak plasma concentration is reached at 1–3 h after oral intake of olaparib, followed by a biphasic decline in plasma concentrations, with a terminal elimination half-life (t ½) of 5–7 h [50]. The main metabolism of olaparib occurs via dehydrogenation and oxidation, with a number of components that were further metabolized by glucuronide or sulphate conjugation [52]. Ang et al. performed a mass balance study of olaparib and found that excretion of the drug occurs mostly via feces (42%) and urine (44%) [53] (Table 1).
Cytochrome P450 (CYP) 3A4 is the main metabolizing enzyme of olaparib; therefore, coadministration of olaparib with strong or moderate CYP3A4 inducers or inhibitors is not recommended. In their dose-escalation study, Fong et al. showed that an increase in olaparib dose led to a linear increase in exposure until olaparib dose levels of 100 mg; exposure did not increase proportionally with an increase in olaparib dose levels higher than 100 mg. The mean apparent volume of distribution is 40 L, with a mean plasma clearance of 4.6 L/h. No severe toxicities were reported, although some mild gastrointestinal toxicities were reported.
Dirix et al. investigated the effect of the CYP3A4 inhibitor itraconazole and the CYP3A4 inducer rifampin on the pharmacokinetics of olaparib. They conducted two phase I studies in patients with advanced solid tumors. Patients received olaparib alone and coadministered with itraconazole or rifampin. Coadministration of olaparib with itraconazole resulted in a statistically significant increase in the relative bioavailability of olaparib, with a peak concentration (C max) treatment ratio of 1.42 (90% confidence interval [CI] 1.33–1.52) and a mean area under the plasma concentration–time curve (AUC) treatment ratio of 2.70 (90% CI 2.44–2.97). A reduction in the mean apparent clearance (CL/F) and apparent volume of distribution (V z /F) was reported. Coadministration with rifampin resulted in a statistically significant reduction in the relative bioavailability of olaparib, with a C max treatment ratio of 0.29 (90% CI 0.24–0.33) and a mean AUC treatment ratio of 0.13 (90% CI 0.11–0.16). These results show that potent CYP3A4 inducers and inhibitors should be avoided during treatment with olaparib [54].
A pharmacokinetic and pharmacodynamic phase I/Ib study of olaparib tablets and carboplatin by Lee et al. was recently published in which 77 patients were treated with olaparib and carboplatin. Patients received either olaparib on days 1–7, carboplatin on day 8, or carboplatin on day 1, followed by olaparib on days 2–8. The clearance of olaparib was increased by approximately 50% when carboplatin was administered 24 h before olaparib, which resulted in a 25% lower AUC from time zero to the last measurable concentration (AUClast; p = 0.046) and a 28% shorter t ½. These results suggest administration of carboplatin prior to olaparib [55].
Fong et al. performed a follow-up study to explore the anti-tumor activity of olaparib in patients with ovarian, primary peritoneal, and fallopian tube cancer. In total, 50 patients, of whom 48 had a germline BRCA1/2 mutation, were treated with olaparib in doses ranging from 40 mg daily to 600 mg twice daily, in a dose-escalation scheme with olaparib monotherapy. Of the 50 patients, 24 had platinum-resistant disease and 13 had platinum-refractory disease. The overall benefit rate was 46%, with a median response duration of 28 weeks. Seventeen patients were treated for more than 6 months [56].
A proof-of-concept study by Audeh et al. [57] confirmed the clinical benefit of olaparib. These researchers treated 55 patients with BRCA1/2 recurrent ovarian cancer, primary peritoneal carcinoma, or fallopian tube carcinoma with olaparib 400 mg twice daily (n = 33) or 100 mg twice daily (n = 24). All patients had recurrence after a previous chemotherapy regimen. The objective response rate (ORR) was significantly better in the 400 mg twice-daily cohort compared with the 100 mg twice-daily cohort (33 vs. 12.5%). All patients except one had at least one adverse event. Most common toxicities were comparable with those observed in previous studies, namely nausea and fatigue. Grade 3 or 4 adverse events were, not as expected, reported slightly more often in the 100 mg twice-daily cohort compared with the 400 mg twice-daily cohort (58 vs. 45%). However, fewer patients discontinued in the 100 mg twice-daily cohort (4%) compared with the 400 mg twice-daily cohort (12%). In terms of anti-tumor activity, the 100 mg twice-daily dose seemed less effective compared with the 400 mg twice-daily dose; however, patients in the lower dosing cohort appeared to have less favorable prognostic factors at the start [57].
The clinical benefit of olaparib was also confirmed in a study by Tutt et al., who also showed a higher ORR in the BRCA-mutated advanced breast cancer group who received 400 mg twice daily compared with the 100 mg twice-daily cohort (41 vs. 22%). Both groups were comparable. All patients were pretreated with at least one chemotherapy regimen. Olaparib also showed activity in patients who were heavily pretreated [58].
As a result of these studies, in 2014 the European Medicines Agency (EMA) approved olaparib monotherapy as treatment for advanced BRCA-mutated ovarian cancer. Following this, the US FDA also approved olaparib for this indication.
Plummer et al. studied the effect of food on the pharmacokinetics of olaparib, and showed that the absorption of olaparib (300 mg once) was lower in the presence of (high fat) food (time to reach C max [T max] delayed by 2.5 h), resulting in a decreased plasma C max of olaparib of 21% [59]. Only a slight increase in olaparib exposure (AUC from time zero to infinity [AUC∞]) was observed, from 43.0 µg h/mL in the fasted state to 45.4 µg h/mL in the fed state.
Besides olaparib, several other PARP inhibitors have been studied. The results of the phase III OlympiAD trial were recently presented. Robson et al. conducted a randomized, open-label, phase III trial in which they compared olaparib monotherapy with standard chemotherapy in patients with BRCA-mutated, HER2-negative, metastatic breast cancer. Patients were not allowed to have received more than two previous lines of chemotherapy for metastatic disease. They received olaparib (300 mg twice daily) or standard ‘physician’s choice’ chemotherapy (capecitabine, eribulin or vinorelbine). In total, 302 patients received treatment, of whom 205 were assigned to the olaparib cohort. Median progression-free survival was significantly longer in the olaparib group than in the standard therapy group (7.0 vs. 4.2 months; p < 0.001), while the response rate was 59.9% in the olaparib group and 28.8% in the standard-therapy group [60].
5.2 Veliparib
Veliparib is also an oral PARP inhibitor. Rugo et al. investigated the combination of veliparib and carboplatin in early breast cancer in the neoadjuvant setting. Patients were randomized to receive either paclitaxel monotherapy or paclitaxel and a combination of veliparib and carboplatin. In both treatment arms, this was followed by four cycles of doxorubicin and cyclophosphamide. The estimated rates of pCR in the TN breast tumors were 51% in the paclitaxel–veliparib–carboplatin group and 26% in the control group. Adding the combination of veliparib-carboplatin to standard chemotherapy in patients with TNBC leads to an increase in the pCR rate compared with standard therapy [61].
Mizugaki et al. conducted a phase I trial to investigate the pharmacokinetics of veliparib in combination with carboplatin and paclitaxel in patients with non-small cell lung cancer (NSCLC), and showed that the addition of carboplatin and paclitaxel had no significant effect on veliparib T max, dose-normalized C max, or dose-normalized AUC. There was also no evidence of an effect of veliparib on the pharmacokinetics of paclitaxel and carboplatin [62].
Nuthalapati et al. conducted a mass balance study of veliparib in subjects with non-hematologic malignancies, and reported pharmacokinetic parameters of veliparib and its metabolite M8 for different doses. Veliparib was rapidly absorbed after oral dosing, with a median T max of 1 h; for its metabolite M8, median T max was 2 h. The systemic exposure of veliparib increased proportional to dose in the dose ranges of 10–80 mg twice daily (this was also seen for the M8 metabolite). Renal elimination of veliparib seemed to be independent of the veliparib dose, and seems to be the major route of elimination of veliparib [63]. Veliparib single-agent therapy has not often been investigated. Coleman et al. performed a multicenter, open-label, phase II trial with veliparib monotherapy and showed activity of the single agent in patients with BRCA-mutated epithelial ovarian, fallopian tube, or primary peritoneal cancer. An ORR of 26% was reached. This was the first phase II study with veliparib monotherapy [64].
5.3 Niraparib
Niraparib is a potent oral PARP-1 and PARP-2 inhibitor with a half maximum inhibitory concentration (IC50) of 3.8 nmol/L for PARP-1 and 2.1 nmol/L for PARP-2 [65]. Sandhu et al. conducted a phase I, dose-escalation study in which 100 patients were enrolled. The AUC was proportional to the dose increase. Absorption was rapid, with a mean plasma concentration peak 3–4 h after a dose, followed by biphasic decrease and a mean t ½ of 36.4 h (range 32.8–46.0). Two of four BRCA-mutated breast cancer cases reached partial responses confirmed by Response Evaluation Criteria In Solid Tumors (RECIST) [66].
In a human mass balance study of 14C-niraparib, Van Andel et al. showed that both renal and hepatic pathways are involved in the excretion of niraparib and its metabolites. The mean total radioactivity recovered in feces and urine was 86.3% (71.1–91.0%) of the total administered dose, of which 38.8% (28.3–47.0%) was recovered in feces and 47.5% (33.4–60.2%) in urine. The elimination of 14C-niraparib was biphasic and slow, with a T ½ in plasma of 92.5 h, on average. Van Andel et al. were able to detect two major metabolites: M1 (amide hydrolyzed niraparib) and the glucuronide of M1 [67]. Very recently, the FDA approved niraparib for the maintenance treatment of adult patients with recurrent high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who were in complete or partial response to platinum-based chemotherapy.
5.4 Differentiation
Olaparib is a potent PARP-1, PARP-2 and PARP-3 inhibitor, whereas niraparib and veliparib are inhibitors of PARP-1 and PARP-2. Phase I trials showed rapid absorption of both niraparib and olaparib. The peak plasma concentration of olaparib seems to have been reached earlier for olaparib (1–3 h) than for niraparib (3–4 h). The decrease in plasma concentration of niraparib is five to seven times slower compared with olaparib. Mizugaki et al. showed that veliparib in the recommended phase II dose (120 mg twice daily) has a peak plasma concentration comparable to niraparib (3.3 h) [62]. Both niraparib and olaparib are eliminated via urine and feces, whereas elimination of veliparib occurs mainly by urine.
6 Combination Therapy
PARP inhibitors have been studied as monotherapy and in combination with radiotherapy or cytotoxic chemotherapy. The putative benefit of combining PARP inhibitors with cytotoxic chemotherapy or radiotherapy is reaching improved efficacy; however, more severe toxicity has been seen in combination trials with chemotherapy and radiotherapy. PARP inhibitors have been studied in several combination studies with cytotoxic agents such as platinum compounds. Oza et al. assessed the tolerability and efficacy of olaparib in combination with chemotherapy followed by olaparib monotherapy compared with chemotherapy alone in patients with high-grade serous ovarian cancer. Patients were randomized between the combination of olaparib (200 mg twice daily) plus paclitaxel (175 mg/m2, administered intravenously on day 1) and carboplatin (AUC 4 min·mg/mL, administered intravenously on day 1), followed by olaparib monotherapy (400 mg twice daily) until progression, or to paclitaxel (175 mg/m2 on day 1) and carboplatin (AUC 6 min·mg/mL, on day 1) not followed by other chemotherapy. Progression-free survival was significantly longer in the olaparib plus chemotherapy group (median 12.2 months, 95% CI 9.7–15.0) than in the chemotherapy-alone group (median 9.6 months, 95% CI 9.1–9.7; hazard ratio [HR] 0.51, 95% CI 0.34–0.77; p = 0.0012), especially in patients with BRCA mutations (HR 0.21, 95% CI 0.08–0.55; p = 0.0015). Adverse events that were more common in the combination group compared with the chemotherapy group alone were alopecia (60 [74%] of 81 vs. 44 [59%] of 75), nausea (56 [69%] vs. 43 [57%]), neutropenia (40 [49%] vs. 29 [39%]), diarrhea (34 [42%] vs. 20 [27%]), headache (27 [33%] vs. 7 [9%]), peripheral neuropathy (25 [31%] vs. 14 [19%]), and dyspepsia (21 [26%] vs. 9 [12%]); most were of mild-to-moderate intensity [68].
Del Conte et al. studied the combination of olaparib and liposomal doxorubicin in patients with advanced solid tumors. Patients received, either continuously (days 1–28) or intermittently (days 1–7), olaparib plus liposomal doxorubicin (40 mg/m2, day 1). The recommended dose was found after dose-escalation of olaparib in seven cohorts (50–400 mg twice daily). It was shown that the C max and AUC from time zero to 10 h (AUC10) of olaparib increased with dose and that the olaparib concentration tended to be higher in the presence of liposomal doxorubicin. During the 28 days of treatment, the minimum plasma concentrations (C min) were maintained, and the most common related toxicities (any grade) were nausea and stomatitis [69].
Van der Noll et al. demonstrated that continuous long-term daily olaparib was safe and tolerable, with manageable side effects and promising anti-tumor effects. These patients (10 with breast cancer, 9 with ovarian cancer, and 2 with fallopian tube cancer) received olaparib monotherapy after treatment with olaparib combined with carboplatin or paclitaxel. The median treatment duration with single-agent olaparib was 52 weeks (7–113) [70].
Another potential combination therapy of PARP inhibitors could be combination with Wee1 inhibitors. Wee1 is a protein kinase that regulates the G2 checkpoint and prevents entry to mitosis in response to DNA damage [71]. Cells with a defective p53 expression are not able to arrest the cell cycle in the G1 phase in order to repair damaged DNA. These cells rely on the G2 checkpoint of the cell cycle for DNA repair [72]. AZD1775 (formerly MK-1775) is a specific inhibitor of the Wee1 kinase. Previous studies have shown a promising safety profile and anti-tumor activity of AZD1775 administered with cytotoxics such as gemcitabine, cisplatin or carboplatin [73, 74]. Karnak et al. performed a study to evaluate the radiosensitization of the combination of the Wee1 inhibitor AZD1775 in combination with olaparib. Their hypothesis was that Wee1 and PARP inhibitors together would give more radiosensitization than either of them alone. They treated pancreatic cells with AZD1775 and olaparib and found that the combination of these agents significantly increased radiosensitivity in these cell lines compared with Wee1 or PARP inhibition alone [75]. Wee1 inhibition could sensitize cells to PARP inhibition through abrogation of the G2 checkpoint and inhibition of HR repair. The combination of these agents resulted in more unrepaired DNA, therefore resulting in cell death. Approximately >80% of the TNBCs have a TP53 mutation, therefore they may be highly sensitive to this combination regimen. Further studies are warranted to investigate this combination in clinical trials.
7 Pharmacodynamic Assays
Pharmacodynamic assay methodologies are designed to determine the effect of the drug on its target [76]. The optimal dose and duration of therapy could be determined, thereby supporting clinical decision making. There are several pharmacodynamic assays that measure PARP activity in tumor cells and peripheral blood mononuclear cells (PBMCs) or lymphocytes [77, 78]. PARP plays a role in the repair of SSBs in the DNA, and produces poly (ADP-ribose) polymers (PARs). Inhibition of PARP is thought to decrease PAR levels. Using an ELISA method, Ji et al. validated a pharmacodynamic assay that quantified PAR levels in both tumor cells and PBMCs [78], and applied this pharmacodynamic assay to a clinical trial of ABT-888.
Kummar et al. performed a phase 0 clinical trial of the PARP inhibitor ABT-888 (veliparib) in patients with advanced malignancies, and determined PAR levels in PBMCs and tumor samples after administration of a single dose of ABT-888. An immunoassay with purified monocloncal antibody to PAR as the capture reagent, and rabbit anti-PAR anti-serum as the detecting agent, were used. A statistically significant inhibition of PAR levels in both tumors and PBMCs was observed after a single dose of ABT-888 [79]. However, PAR levels in PBMCs can be very low, which makes quantification of PAR levels difficult [78, 79].
De Haan et al. aimed to develop a clinically applicable pharmacodynamic assay for quantification of PAR levels and PAR reduction upon PARP inhibition in PBMCs [80]. PBMCs were isolated from the blood of healthy volunteers and NSCLC patients before, during and after treatment with chemoradiation and olaparib. Low levels of PAR are based on low levels of endogenous DNA damage. Therefore, in that study, DNA damage was induced by ex vivo irradiation of the PBMCs. Radiation resulted in an increase of PAR levels in a dose-dependent and linear manner. Another important step in the assay was incubation on ice after irradiation, which resulted in improved PAR signal strength. Clinical studies may benefit from this new assay due to increased sensitivity and the opportunity to correlate the individual patient pharmacodynamic values with individual PARP inhibitor drug response. As a result, more individualization of treatment could be applied.
8 Conclusions
This review summarizes PARP inhibitor treatment of TNBC, and also focuses on patient selection, biomarkers, combination therapy and pharmacodynamic assays.
TNBC has several characteristics that make treatment challenging; it tends to be aggressive and has a high recurrence rate and poor 5-year prognosis compared with other types of breast cancer. Mutations in the BRCA1/2 protein lead to a more error-prone repair pathway due to the function of these genes in the repair of DNA DSBs. TNBC often has similarities with tumors that harbor a BRCA1 mutation. Breast cancer that harbors a BRCA1 or 2 mutation is characterized by a specific pattern of DNA gains and losses in an aCGH assay. This BRCA1/2-likeCGH pattern could help in the selection of patients who may benefit from high-dose DNA DSB-inducing agents.
Six unique subtypes of TNBC have been identified. BL-1 is the most well-known subtype, and has shown a high incidence of BRCA1 methylation and demonstrated similarities to BRCA1-mutated tumors. TP53 is a potential biomarker for patients with TNBC. Since more than 80% of TNBCs have a TP53 mutation, the combination of the Wee1 inhibitor AZD1775 with a PARP inhibitor could be promising.
Given that basal-like TNBC shows similarities with BRCA1-mutated cells, PARP inhibition could be a treatment option for these patients. The combination of PARP inhibition and BRCA1- or 2-mutated tumors shows synthetic lethality, leading to cell death. Olaparib is the most well-known PARP inhibitor and has shown manageable side effects in both the short- and long-term, and promising anti-tumor activity has been demonstrated. Combining olaparib with cytotoxic agents or radiotherapy reaches more efficacy than olaparib monotherapy. Individualization of olaparib treatment is possible due to the use of pharmacodynamic assays, which are able to measure PARP activity in tumor cells and PBMCs. Individual patient pharmacodynamic values could be correlated with the clinical parameters to determine whether the dose has to be adjusted.
Currently, PARP inhibitors are studied in several combination schedules with cytotoxic agents and radiotherapy. As well as in patients with BRCA mutation, PARP inhibition is being studied in patients with the BRCAness phenotype, which could lead to a broader application of PARP inhibitors. Since 30% of the sporadic tumors have the BRCAness phenotype, clinical trials must show whether there is an increased anti-tumor effect combining these agents, with manageable side effects. Individualization of treatment plays a role in daily practice more and more. When pharmacodynamic assays are generally applied in treatment with PARP inhibitors, under- and overdosing could be prevented; however, this concept needs prospective clinical validation.
References
World Health Organization. Breast cancer, global health estimates. Geneva: World Health Organization; 2013.
Liu M, Li Z, Yang J, Jiang Y, Chen Z, Ali Z, et al. Cell-specific biomarkers and targeted biopharmaceuticals for breast cancer treatment. Cell Prolif. 2016;49(4):409–20.
Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805(1):105–17.
Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16(5):335–43.
Jia LY, Shanmugam MK, Sethi G, Bishayee A. Potential role of targeted therapies in the treatment of triple-negative breast cancer. Anticancer Drugs. 2016;27(3):147–55.
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig. 2011;121(7):2750–67.
Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81.
Lips EH, Mulder L, Hannemann J, Laddach N, Vrancken Peeters MT, van de Vijver MJ, et al. Indicators of homologous recombination deficiency in breast cancer and association with response to neoadjuvant chemotherapy. Ann Oncol. 2011;22(4):870–6.
Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31(6):955–60.
Bunting SF, Callen E, Kozak ML, Kim JM, Wong N, Lopez-Contreras AJ, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. 2012;46(2):125–35.
Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–95.
Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–15.
Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–8.
Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP. The role of BRCA1 in the cellular response to chemotherapy. J Natl Cancer Inst. 2004;96(22):1659–68.
Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA. 2007;104(29):12117–22.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105(44):17079–84.
Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci. 2010;31(8):372–80.
Lewin R, Sulkes A, Shochat T, Tsoref D, Rizel S, Liebermann N, et al. Oncotype-DX recurrence score distribution in breast cancer patients with BRCA1/2 mutations. Breast Cancer Res Treat. 2016;157(3):511–6.
Sharma P, Klemp JR, Kimler BF, Mahnken JD, Geier LJ, Khan QJ, et al. Germline BRCA mutation evaluation in a prospective triple-negative breast cancer registry: implications for hereditary breast and/or ovarian cancer syndrome testing. Breast Cancer Res Treat. 2014;145(3):707–14.
Murphy CG, Moynahan ME. BRCA gene structure and function in tumor suppression: a repair-centric perspective. Cancer J. 2010;16(1):39–47.
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9.
Andreopoulou E, Schweber SJ, Sparano JA, McDaid HM. Therapies for triple negative breast cancer. Expert Opin Pharmacother. 2015;16(7):983–98.
Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J Clin Oncol. 2015;33(1):13–21.
Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764–73.
Byrski T, Dent R, Blecharz P, Foszczynska-Kloda M, Gronwald J, Huzarski T, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012;14(4):R110.
Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28(3):375–9.
Bal A, Verma S, Joshi K, Singla A, Thakur R, Arora S, et al. BRCA1-methylated sporadic breast cancers are BRCA-like in showing a basal phenotype and absence of ER expression. Virchows Arch. 2012;461(3):305–12.
Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006;8(4):R38.
Wei M, Grushko TA, Dignam J, Hagos F, Nanda R, Sveen L, et al. BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res. 2005;65(23):10692–9.
Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013;108(10):2172–7.
Metzger-Filho O, Tutt A, de Azambuja E, Saini KS, Viale G, Loi S, et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol. 2012;30(15):1879–87.
Lu W, Wang X, Lin H, Lindor NM, Couch FJ. Mutation screening of RAD51C in high-risk breast and ovarian cancer families. Fam Cancer. 2012;11(3):381–5.
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39(2):165–7.
Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, et al. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res. 2013;32:95.
Lehmann BD, Pietenpol JA, Tan AR. Triple-negative breast cancer: molecular subtypes and new targets for therapy. In: American Society of Clinical Oncology educational book/ASCO American Society of Clinical Oncology meeting. 2015. p. e31–9.
Chakravarthy AB, Kelley MC, McLaren B, Truica CI, Billheimer D, Mayer IA, et al. Neoadjuvant concurrent paclitaxel and radiation in stage II/III breast cancer. Clin Cancer Res. 2006;12(5):1570–6.
Bauer JA, Chakravarthy AB, Rosenbluth JM, Mi D, Seeley EH, De Matos Granja-Ingram N, et al. Identification of markers of taxane sensitivity using proteomic and genomic analyses of breast tumors from patients receiving neoadjuvant paclitaxel and radiation. Clin Cancer Res. 2010;16(2):681–90.
Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans V, et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;366(9503):2087–106.
Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013;19(19):5533–40.
Holstege H, Horlings HM, Velds A, Langerod A, Borresen-Dale AL, van de Vijver MJ, et al. BRCA1-mutated and basal-like breast cancers have similar aCGH profiles and a high incidence of protein truncating TP53 mutations. BMC Cancer. 2010;10:654.
Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26(14):2126–32.
Hill SJ, Clark AP, Silver DP, Livingston DM. BRCA1 pathway function in basal-like breast cancer cells. Mol Cell Biol. 2014;34(20):3828–42.
Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25(43):5846–53.
Joosse SA, van Beers EH, Tielen IH, Horlings H, Peterse JL, Hoogerbrugge N, et al. Prediction of BRCA1-association in hereditary non-BRCA1/2 breast carcinomas with array-CGH. Breast Cancer Res Treat. 2009;116(3):479–89.
Waddell N, Arnold J, Cocciardi S, da Silva L, Marsh A, Riley J, et al. Subtypes of familial breast tumours revealed by expression and copy number profiling. Breast Cancer Res Treat. 2010;123(3):661–77.
Tirkkonen M, Johannsson O, Agnarsson BA, Olsson H, Ingvarsson S, Karhu R, et al. Distinct somatic genetic changes associated with tumor progression in carriers of BRCA1 and BRCA2 germ-line mutations. Cancer Res. 1997;57(7):1222–7.
Jonsson G, Naylor TL, Vallon-Christersson J, Staaf J, Huang J, Ward MR, et al. Distinct genomic profiles in hereditary breast tumors identified by array-based comparative genomic hybridization. Cancer Res. 2005;65(17):7612–21.
Vollebergh MA, Lips EH, Nederlof PM, Wessels LF, Schmidt MK, van Beers EH, et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann Oncol. 2011;22(7):1561–70.
Vollebergh MA, Lips EH, Nederlof PM, Wessels LF, Wesseling J, Vd Vijver MJ, et al. Genomic patterns resembling BRCA1- and BRCA2-mutated breast cancers predict benefit of intensified carboplatin-based chemotherapy. Breast Cancer Res. 2014;16(3):R47.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Clarkson-Jones J PC, Sarda S, et al. Human biotransformation of olaparib (AZD2281) an oral poly(ADP-ribose) polymerase (PARP) inhibitor [abstract no. 417]. In: 22nd EORTC-NCI-AACR symposium on molecular targets and cancer therapeutics. 2010.
Ang JE C-JJ, Swaisland H, et al. A mass balance study to investigate the metabolism, excreation and pharmacokinetics of [14]-olaparib (AZD2281) in patients with advanced solid tumours refractory to standard treatments [abstract no. 405]. In: 22nd EORTC-NCI-AACR symposium on molecular targets and cancer therapeutics. 2010.
Dirix L, Swaisland H, Verheul HM, Rottey S, Leunen K, Jerusalem G, et al. Effect of itraconazole and rifampin on the pharmacokinetics of olaparib in patients with advanced solid tumors: results of two phase I open-label studies. Clin Ther. 2016;38(10):2286–99.
Lee JM, Peer CJ, Yu M, Amable L, Gordon N, Annunziata CM, et al. Sequence-specific pharmacokinetic and pharmacodynamic phase I/Ib study of olaparib tablets and carboplatin in women’s cancer. Clin Cancer Res. 2017;23(6):1397–406.
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–9.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–51.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44.
Plummer R, Swaisland H, Leunen K, van Herpen CM, Jerusalem G, De Greve J, et al. Olaparib tablet formulation: effect of food on the pharmacokinetics after oral dosing in patients with advanced solid tumours. Cancer Chemother Pharmacol. 2015;76(4):723–9.
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–33. doi:10.1056/NEJMoa1706450.
Rugo HS, Olopade OI, DeMichele A, Yau C, van ‘t Veer LJ, Buxton MB, et al. Adaptive randomization of veliparib–carboplatin treatment in breast cancer. N Engl J Med. 2016;375(1):23–34.
Mizugaki H, Yamamoto N, Nokihara H, Fujiwara Y, Horinouchi H, Kanda S, et al. A phase 1 study evaluating the pharmacokinetics and preliminary efficacy of veliparib (ABT-888) in combination with carboplatin/paclitaxel in Japanese subjects with non-small cell lung cancer (NSCLC). Cancer Chemother Pharmacol. 2015;76(5):1063–72.
Nuthalapati S, Munasinghe W, Giranda V, Xiong H. Clinical pharmacokinetics and mass balance of veliparib in combination with temozolomide in subjects with nonhematologic malignancies. Clin Pharmacokinet. 2017. doi:10.1007/s40262-017-0547-z.
Coleman RL, Sill MW, Bell-McGuinn K, Aghajanian C, Gray HJ, Tewari KS, et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation. An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2015;137(3):386–91.
Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem. 2009;52(22):7170–85.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–92.
van Andel L, Zhang Z, Lu S, Kansra V, Agarwal S, Hughes L, et al. Human mass balance study and metabolite profiling of 14C-niraparib, a novel poly(ADP-Ribose) polymerase (PARP)-1 and PARP-2 inhibitor, in patients with advanced cancer. Investig New Drugs. 2017. doi:10.1007/s10637-017-0451-2.
Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(1):87–97.
Del Conte G, Sessa C, von Moos R, Vigano L, Digena T, Locatelli A, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer. 2014;111(4):651–9.
van der Noll R, Marchetti S, Steeghs N, Beijnen JH, Mergui-Roelvink MW, Harms E, et al. Long-term safety and anti-tumour activity of olaparib monotherapy after combination with carboplatin and paclitaxel in patients with advanced breast, ovarian or fallopian tube cancer. Br J Cancer. 2015;113(3):396–402.
Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle. 2013;12(19):3159–64.
Leijen S, Beijnen JH, Schellens JH. Abrogation of the G2 checkpoint by inhibition of Wee-1 kinase results in sensitization of p53-deficient tumor cells to DNA-damaging agents. Curr Clin Pharmacol. 2010;5(3):186–91.
Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34(36):4371–80.
Leijen S, van Geel RMJM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34(36):4354–61.
Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res. 2014;20(19):5085–96.
Kinders RJ, Hollingshead M, Khin S, Rubinstein L, Tomaszewski JE, Doroshow JH, et al. Preclinical modeling of a phase 0 clinical trial: qualification of a pharmacodynamic assay of poly (ADP-ribose) polymerase in tumor biopsies of mouse xenografts. Clin Cancer Res. 2008;14(21):6877–85.
Bundred N, Gardovskis J, Jaskiewicz J, Eglitis J, Paramonov V, McCormack P, et al. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: a phase I multicentre trial in patients scheduled for elective breast cancer surgery. Investig New Drugs. 2013;31(4):949–58.
Ji J, Kinders RJ, Zhang Y, Rubinstein L, Kummar S, Parchment RE, et al. Modeling pharmacodynamic response to the poly(ADP-ribose) polymerase inhibitor ABT-888 in human peripheral blood mononuclear cells. PLoS One. 2011;6(10):e26152.
Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, et al. Phase 0 clinical trial of the poly(ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27(16):2705–11.
de Haan R, Pluim D, van Triest B, van den Heuvel M, Peulen H, van Berlo D, George J, Verheij M, Schellens JHM, Vens C. Development and validation of a sensitive pharmacodynamic method for individualized treatment with PARP inhibitors. Radiother Oncol. 2016. (in press).
Mostafa NM, Chiu YL, Rosen LS, Bessudo A, Kovacs X, Giranda VL. A phase 1 study to evaluate effect of food on veliparib pharmacokinetics and relative bioavailability in subjects with solid tumors. Cancer Chemother Pharmacol. 2014;74(3):583–91.
Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. An update on PARP-inhibitors: moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41.
Wiegand R, Wu J, Sha X, LoRusso P, Li J. Simultaneous determination of ABT-888, a poly (ADPribose) polymerase inhibitor, and its metabolite in human plasma by liquid chromatography/tandem mass spectometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:333–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was provided for the preparation of this article.
Conflicts of Interest
Jill J. J. Geenen, Jan H. M. Schellens and Jos H. Beijnen declare no conflicts of interest. Sabine C. Linn received a research grant and study drug (olaparib) for the REVIVAL study (NCT02810743), and is a member of the advisory board for olaparib in breast cancer (paid to institution), Dr. Linn also has a patent (means and methods for molecular classification of BRCA-like breast and/or ovarian cancer).
Rights and permissions
About this article

Cite this article
Geenen, J.J.J., Linn, S.C., Beijnen, J.H. et al. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin Pharmacokinet 57, 427–437 (2018). https://doi.org/10.1007/s40262-017-0587-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0587-4