
Abstract
Vonoprazan fumarate (Takecab®) is a first-in-class potassium-competitive acid blocker that has been available in the market in Japan since February 2015. Vonoprazan is administered orally at 20 mg once daily for the treatment of gastroduodenal ulcer, at 20 and 10 mg once daily for the treatment and secondary prevention of reflux esophagitis, respectively, at 10 mg once daily for the secondary prevention of low-dose aspirin- or non-steroidal anti-inflammatory drug-induced peptic ulcer, and at 20 mg twice daily in combination with clarithromycin and amoxicillin for the eradication of Helicobacter pylori. It inhibits H+,K+-ATPase activities in a reversible and potassium-competitive manner with a potency of inhibition approximately 350 times higher than the proton pump inhibitor, lansoprazole. Vonoprazan is absorbed rapidly and reaches maximum plasma concentration at 1.5–2.0 h after oral administration. Food has minimal effect on its intestinal absorption. Oral bioavailability in humans remains unknown. The plasma protein binding of vonoprazan is 80 % in healthy subjects. It distributes extensively into tissues with a mean apparent volume of distribution of 1050 L. Being a base with pKa of 9.6 and with acid-resistant properties, vonoprazan is highly concentrated in the acidic canaliculi of the gastric parietal cells and elicited an acid suppression effect for longer than 24 h after the administration of 20 mg. The mean apparent terminal half-life of the drug is approximately 7.7 h in healthy adults. Vonoprazan is metabolized to inactive metabolites mainly by cytochrome P450 (CYP)3A4 and to some extent by CYP2B6, CYP2C19, CYP2D6, and SULT2A1. A mass balance study showed that 59 and 8 % of the orally administered radioactivity was recovered in urine as metabolites and in an unchanged form, respectively, indicating extensive metabolism. Genetic polymorphism of CYP2C19 may influence drug exposure but only to a clinically insignificant extent (15–29 %), according to the population pharmacokinetic study performed in Japanese patients. When vonoprazan was co-administered with clarithromycin, the mean AUC from time 0 to time of the next dose (dosing interval) of vonoprazan and clarithromycin were increased by 1.8 and 1.5 times, respectively, compared with the corresponding control values, indicating mutual metabolic inhibition. The mean area under the curve from time zero to infinity obtained from patients with severe liver and renal dysfunction were elevated by 2.6 and 2.4 times, respectively, compared with healthy subjects, with no significant changes in plasma protein binding. Vonoprazan increases intragastric pH above 4.0 as early as 4 h after an oral dose of 20 mg, and the extensive anti-secretory effect is maintained up to 24 h post-dose. During repeated dosing of 20 mg once daily, the 24-h intragastric pH >4 holding time ratios were 63 and 83 % on days 1 and 7, respectively. Because vonoprazan elicited a more extensive gastric acid suppression than the proton pump inhibitor, lansoprazole, it also gave rise to two to three times greater serum gastrin concentrations as compared with lansoprazole. In pre-approval clinical studies for the treatment of acid-related disorders, mild to moderate adverse drug reactions (mostly constipation or diarrhea) occurred at frequencies of 8–17 %. Neither severe liver toxicity nor neuroendocrine tumor has been reported in patients receiving vonoprazan.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Vonoprazan is a first-in-class potassium-competitive blocker. |
It inhibits the H+,K+-ATPase-mediated gastric acid secretion in a reversible and potassium-competitive manner. |
It possesses approximately 350 times more potent inhibitory effects than the proton pump inhibitor, lansoprazole, in in vitro experiments. |
It increased the gastric pH above 4.0 as early as 4 h after a single oral dose of 20 mg and maintained the gastric pH above 4.0 until 24 h post-dose. |
It undergoes substantial metabolic elimination but the influence of a genetic polymorphism is limited. |
1 Introduction
Acid-related diseases such as gastroduodenal ulcer and gastroesophageal reflux disease cause substantial morbidity and are an important public health issue. The mainstay of treatment for these disorders has been suppression of gastric acid secretion. Historically, antacids, histamine H2-receptor antagonists, and muscarinic receptor antagonists were used until proton pump inhibitors (PPIs) became available in the late 1980s. Having more potent acid-suppressing effects and better tolerability than the above-named drugs, PPIs have been considered the drug of choice for most patients with acid-related disorders in the last two decades [1]. In addition, with increasing recognition that Helicobacter pylori is associated with recurrence of peptic ulcer disease and is the etiology of H. pylori-positive, mucosa-associated lymphatic tissue lymphoma, immune thrombocytopenia, and possibly gastric adenocarcinoma, eradication therapy of H. pylori has been performed widely [2]. One of the PPIs has been included in the combination chemotherapy with amoxicillin and clarithromycin, aiming to boost the antimicrobial effect against H. pylori [2].
Because PPIs revolutionized the pharmacotherapy of acid-related disorders so successfully, many clinicians doubted whether an anti-secretory drug more powerful than PPIs would ever be developed. However, industry researchers continued their endeavors in the ensuing two decades to develop such a drug. Because PPIs have a relatively slow onset of action, three to five dose cycles have to be administered before they exhibit maximum anti-secretory effects [3]. In addition, PPIs may not be able to fully suppress night-time acid breakthrough [4]. As a result, approximately one-third of patients with gastroesophageal reflux disease receiving PPIs still need supplemental antacids for symptomatic relief or for enduring residual symptoms [5]. Clinical studies revealed that PPI therapy may not be sufficient for the extensive pH control needed to prevent rebreeding after endoscopic hemostasis in some patients [6]. Furthermore, because the pharmacokinetics of many PPIs are subject to the influence of genetic polymorphisms of cytochrome P450 (CYP)2C19, the standard dose of PPIs may not provide sufficient anti-secretory effects in extensive metabolizers of CYP2C19 for the eradication of H. pylori [7]. Finally, PPIs require an acid-resistant formulation [3], otherwise they are transformed to the active protonated sulfonamides in the gastric lumen before reaching the sites of action that are located in the secretory canaliculi of the gastric parietal cells.
An alternative strategy exists for searching an inhibitor of H+,K+-ATPase. When protons are transported by H+,K+-ATPase from the cytoplasm to the canalicular space across the apical membrane of the parietal cell, an equal amount of K+ ions are counter transported into the cytoplasm of the parietal cells so that the total process is electrically balanced [8]. As a result, the activity of H+,K+-ATPase in the parietal cells is regulated by the availability of extracellular K+. Studies on the molecular mechanisms of H+,K+-ATPase led to the discovery of a new class of anti-secretory agent, potassium-competitive acid blocker (P-CAB). This class of drugs exhibit their anti-secretory effects by competitively blocking the availability of K+ for the H+,K+-ATPase [8]. Following the development of a prototype P-CAB, SCH28080, many candidate compounds (e.g., TAK-438, BY841, SK&F 96067, AZD0865, CS-526, and YH1885) were developed, but some of them were withdrawn after clinical trials owing to the short duration of action or hepatic toxicity [8]. YH1885 (revaprazan) was launched in Korea in 2007 for the treatment of gastroduodenal ulcer and gastritis, whereas only limited data are available for its pharmacokinetics and details of relevant clinical trials are available only in Korean [9, 10]. Vonoprazan fumarate (TAK-438, Takecab®) is another P-CAB that was approved by the Japanese Ministry of Health, Labour and Welfare in December 2014 and was launched in February 2015 for the treatment of acid-related disorders and as an adjunct to H. pylori eradication. Vonoprazan is a first-in-class P-CAB of which pharmacokinetic and pharmacodynamic data are characterized in Caucasian and Asians [11]. In addition, its efficacy and safety for the treatment of acid-related diseases has been verified as compared to a PPI, lansoprazole, in well-designed controlled clinical trials. The summarized data of individual clinical trials were presented at the Digestive Disease Week in 2014 and 2015 and published in abstract form subsequently [12–18]. A brief summary of therapeutic trials of vonoprazan was reported recently [19], but more detailed information is available at this moment only in Japanese [20–22]. The present article is the first review article on the pharmacokinetics and pharmacodynamics of vonoprazan using information published as of July, 2015 in English or Japanese.
2 Physicochemical Characteristics and Relevant Pharmacologic Properties
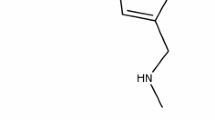
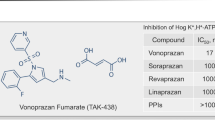
Vonoprazan is an acid-resistant pyrrole derivative 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine fumarate (TAK-438) developed by Takeda Pharmaceutical Company Ltd. as a P-CAB [23] (Fig. 1). Because vonoprazan has a greater point-positive charge (pKa 9.06) than other P-CABs with different chemical structures (such as SCH28080 with pKa 5.6), it is accumulated at higher concentrations in the canalicular space of the gastric parietal cells compared with the previous P-CABs [24]. The drug is bound to H+,K+-ATPase in a strictly K+-competitive and reversible manner with a Ki of 10 nM at pH 7.0 [24]. Vonoprazan is 10 and 350 times more potent in H+,K+-ATPase inhibitory activity than a prototype P-CAB, SCH28080, and a PPI, lansoprazole, respectively, in porcine gastric microsomes at pH 6.5; the half maximal inhibitory concentrations (IC50) were 0.019 nM, 0.14 nM, and 7.6 μM, respectively [24]. In addition, the rate of dissociation of vonopazan from isolated ATPase is slower (half-life: 7.5 h in 20 mM KCL, close to physiological concentrations in stimulated gastric juice at pH 7.0) than other P-CABs (2 min for SCH28080 in 10 mM KCL). These in vitro pharmacological properties may account for the more potent and longer lasting anti-secretory effects of vonopazan than lansoprazole observed in in vivo animal experiments [25].

Chemical structure of vonoprazan (reproduced from [9] with permission from the publisher, Adis International Ltd.)
3 Pharmacokinetics
3.1 Healthy Subjects
3.1.1 Absorption
Being acid resistant, vonoprazan can be given as a rapid-release formulation. The pharmacokinetics of the drug after oral administration were studied in Japan (n = 60) and the UK (n = 48) [11]. While no model-based pharmacokinetic analysis has been performed on the data, the intestinal absorption of vonoprazan appears rapid. After single oral doses of the drug at 10, 15, 20, 30, and 40 mg under fasting conditions, the maximum plasma concentration (C max) increased from approximately 10 to 60 ng/mL in a largely dose-proportional manner in both study populations, with time to reach C max of 1.5–2.0 h [11]. The data obtained during repeated oral dosing of the drug at 10, 15, 20, 30, and 40 mg once daily for 7 days showed that the drug accumulation was almost completed by the third day with accumulation factors for C max and area under the curve from time zero to infinity (AUC0–inf) at the respective doses ranging from 1.14 to 1.32 [11]. Dose proportionality for C max and AUC0–inf was observed on the first day and seventh day in both study populations.
3.1.1.1 Bioavailability
The oral bioavailability of vonoprazan in humans remains unknown because no intravenous pharmacokinetic study has been performed. Nevertheless, a mass balance study performed in healthy subjects after a single oral dose of 15 mg 14C-vonoprazan demonstrated that 67 and 31 % of the total radioactivity administered was recovered by 168 h post-dose in urine and feces, respectively [20]. In addition, the unchanged form of the drug recovered in urine and feces accounted for only 8 and 1 %, respectively, of the total radioactivity administered. Furthermore, the plasma AUC attributable to the unchanged form accounted for only 13 % of the total radioactivity. These data suggest that intestinal absorption of the drug is fair (>67 %) and the drug undergoes extensive metabolism. As the oral bioavailability of vonoprazan is unknown, it remains unclear whether the drug is subject to a substantial first-pass elimination in either the liver or the gut, or combination thereof, in humans. The pharmacokinetics of 14C-vonoprazan after intravenous and oral administration were studied in rats and dogs. The oral bioavailability was 10 % in rats and 52 % in dogs, despite gastrointestinal absorption being >80 % in both species. These data indicate the presence of substantial to moderate first-pass metabolism in these species [20]. Whether substantial metabolism takes place in the intestinal wall and contributes significantly to the first-pass metabolism remains unclear in these species. However, an absorption study performed with perfused jejunum loops of rats showed that 42 % of 14C-vonoprazan administered into the loop was recovered in the portal blood within 2 h, mainly in the unchanged form (>89 %). These findings imply that the gut may not be involved in the substantial metabolism of vonoprazan, at least in rats [20].
3.1.1.2 Effects of Food Ingestion on Gastrointestinal Absorption
The effects of a meal on the pharmacokinetics of vonoprazan were examined in a randomized crossover trial. Twelve healthy subjects took an oral dose of vonoprazan 20 mg before breakfast (fasting) in one study and after breakfast in the other, separated by a washout period of 7 days [20–22]. The ratios of geometric means for C max and area under the curve from time zero to 48 h (AUC0–48h) obtained after a meal to those obtained under fasting conditions were 1.09 [90 % confidence interval (CI) 0.94–1.26] and 1.12 (90 % CI 1.01–1.14), respectively, indicating that the effect of a meal on intestinal absorption of the drug, if any, is clinically insignificant.
3.1.2 Distribution
Because no plasma concentration–time data after an intravenous administration of vonoprazan are available, only apparent volume of distribution for the drug is available at present. In a study performed with 12 healthy men, the mean (±standard deviation [SD]) volume of distribution was 1056 ± 263 L after an oral dose of 20 mg vonoprazan under fasting conditions [20]. Because the bioavailability of vonoprazan is theoretically no less than 8 % (see Sect. 3.1.1.1), the true Vd is estimated to be 84 L or greater. Because vonoprazan is a base with pKa >9.0 [21], it is accumulated in the acidic or neutral secretory canaliculi of the gastric parietal cells in much higher concentrations than in plasma. An experiment performed using rats demonstrated that the mean gastric wall concentrations of vonoprazan was more than 1000 times greater than plasma concentrations at 5 h after an intravenous dose [20]. As described above, vonoprazan is accumulated in the secretory canaliculi more extensively than other P-CABs because it has a greater point-positive charge [24].
3.1.2.1 Plasma Protein Binding
The unbound fractions of vonoprazan in human plasma spiked with the drug to concentrations of 0.1, 1, and 10 μg/mL were 14, 15, and 12 %, respectively [20, 22]. Because plasma vonoprazan concentrations attained after an oral dose approved for the treatment of acid-related diseases (10 or 20 mg once daily) are less than 50 ng/mL [22], plasma protein binding of the drug is unlikely to be saturated even at concentrations well above the therapeutic range. In addition, the drug is bound to both albumin and α1-acid glycoprotein [20].
3.1.3 Elimination
3.1.3.1 Clearance
The mean (±SD) oral clearance of vonoprazan after an oral dose of 20 mg in healthy subjects under fasting conditions was 97.5 ± 30.1 L/h (1.63 ± 0.50 L/min) [20]. Because the oral bioavailability of vonoprazan is unknown, its systemic clearance cannot be calculated. The blood to plasma concentration ratio of the drug was shown to be 0.93–0.93 [20]. The mean apparent terminal half-life obtained from healthy subjects after a single oral dose under fasting conditions was 7.7 h, irrespective of the administration under fasting or after a meal [22].
3.1.3.2 Metabolism
In vitro studies on the metabolism of vonoprazan using human liver microsomes revealed that the drug is metabolized mainly by CYP3A4 and to some extent by CYP2B6, CYP2C19, CYP2D6, and SULT2A1 [20–22]. None of the metabolites are pharmacologically active. Vonoprazan has been shown to have a direct inhibitory effect on CYP2B6 and CYP3A4/5, with IC50 of 16 and 29 μmol/L, respectively. In addition, vonoprazan is known to inhibit the activities of CYP2B6, CYP2C19, and CYP3A4/5 in a time-dependent manner, with IC50 of 3, 13, 10, and 10 μmol/L, respectively [20]. Because the maximum plasma concentrations of the drug after therapeutic oral doses are approximately 100–200 nmol/L, which are far lower than the above IC50 values, vonoprazan may be less likely to have clinically significant inhibitory effects on these CYP isozymes in clinical situations [20]. In addition, the drug has been shown to inhibit the transporter activity of P-glycoprotein (P-gp), with an IC50 of 50 μmol/L. According to the guidelines of drug interaction for industries issued by the US Food and Drug Administration and the European Medicines Agency [26, 27], industries are requested to conduct clinical studies of the drug under development, if maximum drug concentrations in the intestinal lumen are estimated to be 10 times higher than the IC50 for P-gp observed in in vitro experiments. The maximum concentrations of vonoprazan in the intestinal lumen are approximately 230 μmol/L, which exceeds the IC50 for P-gp by only five times. Therefore, it is considered, at least theoretically, that vonoprazan may be less likely to elicit a clinically significant inhibitory effect on P-gp [20]. However, further clinical studies are required to confirm the theoretical considerations for the drug interaction via the inhibition of drug metabolism and P-gp.
3.1.3.3 Renal Excretion
The mean renal clearance of vonoprazan is 6.4 L/h (107 mL/min) in healthy subjects (n = 13) and only 4 % of the oral dose is recovered in urine as the unchanged form [21]. Because the systemic clearance of the drug remains unclear, it may be difficult to conclude whether renal elimination contributes substantially to systemic clearance of the drug based only on the pharmacokinetics of healthy subjects. However, it is obvious that an active transport mechanism is involved in renal elimination of vonoprazan, because renal clearance for the unbound drug (approximately 700 mL/min) apparently far exceeds the glomerular filtration rate (120 mL/min).
3.2 Special Populations
3.2.1 Liver Disease
A clinical study was performed to investigate the influence of liver disease on the pharmacokinetics of vonoprazan after a single oral dose of 20 mg in patients with mild, moderate, and severe liver dysfunction classified as Child–Pugh classes A, B, and C (n = 8 in each group), respectively, compared with healthy subjects (n = 8) [20–22]. The geometric means for C max and AUC0–inf obtained from patients with Child–Pugh classes A, B, and C were greater than the corresponding values obtained from healthy subjects by 1.2 and 1.2, 1.8 and 2.4, and 1.8 and 2.6 times, respectively. No data were available for changes in the half-life. The mean plasma unbound fractions of vonoprazan obtained from patients with mild, moderate, and severe liver dysfunction and healthy controls were 19, 24, 23, and 21 %, respectively, with no significant differences between groups. Collectively, these data are compatible with the idea that the metabolic elimination is substantially involved in the systemic clearance of the drug. The clinical implications of these data remain to be verified. At present, the prescribing information contains no specific recommendations for dosage reduction in patients with severe liver dysfunction [22].
3.2.2 Renal Disease
A pharmacokinetic study was performed in patients with mild, moderate, and severe renal dysfunction as well as those with end-stage renal disease (ESRD) having an estimated glomerular filtration rate (eGFR) of 60–89, 30–59, 15–29 (n = 8 each), and <15 mL/min/1.73 m2 (n = 3), respectively, compared with healthy subjects having an eGFR of >90 mL/min/1.73 m2 (n = 13), after a single oral dose of 20 mg vonoprazan [20, 22]. The geometric mean of C max and AUC0–inf obtained from patients with mild, moderate, and severe renal dysfunction, and ESRD were greater than the corresponding values obtained from healthy controls by 2.3 and 1.7, 1.2 and 1.3, and 2.8 and 2.4, and 1.2 and 1.3 times, respectively. The mean renal clearance (L/h) in patients with mild, moderate, severe, and end-stage renal dysfunction were 5.4, 4.6, 1.8, and 0.5 L/h, respectively, compared with 6.4 L/h in healthy subjects. In addition, the mean percentages of vonoprazan excreted into urine as an unchanged form obtained from the corresponding groups of patients were reduced in association with their renal function to 5.4, 3.6, 2.8, and 0.4 %, respectively, compared with 4.0 % for healthy subjects. Mean plasma unbound fractions of vonoprazan in healthy subjects and patients with mild, moderate, severe, and end-stage renal dysfunction were 21, 21, 23, and 20 %, respectively, with no significant differences between groups. The reason why the AUC of the drug was increased in patients with renal dysfunction despite only less than 5 % of the unchanged form of the drug eliminated into urine remains to be studied. The clinical implications of these data remain to be determined. At present, the prescribing information contains no specific recommendations for dosage reduction in patients with severe liver dysfunction [22].
3.2.3 Sex
A pharmacokinetic study performed in healthy men and women (n = 12 each) after a single oral dose of 20 mg demonstrated that the female/male ratio for the geometric means (95 % CI) of C max and AUC0–inf were 0.78 (0.52–1.17) and 0.84 (0.54–1.31), respectively [20]. While there is a trend towards greater oral clearance in women than in men, the difference did not reach a significant level. Supplemental data for sex difference in the pharmacokinetics of the drug are provided in the following section.
3.2.4 CYP2C19 Polymorphism
An exploratory study was performed to measure plasma concentrations of vonoprazan in 60 Japanese patients whose genotypes of CYP2C19 were identified [11]. These subjects took 10–40 mg of vonoprazan once daily for 7 days. No appreciable correlation was found between CYP2C19 genotypes (*1/*1, *1/*2, *1/*3, and *2*2) and mean dose-normalized AUC from time 0 to time of the next dose (dosing interval) (AUC0–tau) at steady state. In contrast, a population pharmacokinetic modeling analysis using the non-linear mixed-effect model software performed on 1751 plasma samples obtained from 592 patients with reflux esophagitis who participated in phase II dose-finding studies revealed that among the variables examined, dose, sex, age, and CYP2C19 were significantly associated with the inter-individual variability of oral clearance of vonoprazan [20]. Specifically, the median values of oral clearance for women obtained by individual post hoc estimation at different doses were 5–24 % greater than the corresponding values for men. The values obtained from patients aged from 65 to 75 years and those aged >75 years were lower than those aged <65 years by 18–25 % and 19–35 %, respectively. The values obtained from patients with CYP2C19 genotypes associated with the poor metabolizer phenotype were 15–29 % lower than those associated with the extensive metabolizer phenotype. Because the magnitudes of the effect caused by these covariates were less than 35 % compared with the population mean, these variables may exert only clinically insignificant influence on the overall exposure of vonoprazan.
3.3 Drug Interaction
3.3.1 Absorption
Theoretically, vonoprazan may attenuate the bioavailability of drugs of which gastrointestinal absorption is susceptible to changes in gastric pH. Indeed, PPIs have been shown to reduce the mean C max and AUC0–24 of atazanavir by 94 and 91 %, respectively, compared with the corresponding values observed for atazanavir alone (for review see [28]). There is a possibility that vonoprazan also interferes with the bioavailability of those drugs to a similar extent as PPIs. At present, no data are available to negate or support such a theoretical concern.
3.3.2 Metabolism
As was discussed in Sect. 3.1.3.2, vonoprazan is a substrate and may be an inhibitor of CYP3A4 and some other CYP isoforms [20]. Because the drug has an approved indication for the eradication of H. pylori by co-administration with clarithromycin, a strong CYP3A inhibitor [26], there is a concern over whether there is a significant drug interaction between vonoprazan and clarithromycin. In this context, a clinical study was performed in healthy subjects to investigate whether co-administration of vonoprazan (20 mg), clarithromycin (400 mg), and amoxicillin (750 mg) twice daily for 7 days alters the pharmacokinetics of the drugs compared with the respective drugs when used alone (control values) [20–22]. This study demonstrated that mean C max and area under the curve from time zero to 12 h (AUC0–12) for vonoprazan increased by 1.9 and 1.8 times on the seventh day, and those for clarithromycin also increased by 1.6 and 1.5 times compared with the respective control values. In contrast, no changes were observed in mean C max and AUC0–12 for amoxicillin. In addition, the pharmacokinetics of vonoprazan after an oral dose of 40 mg once daily was studied before and after co-administration of oral clarithromycin 500 mg twice daily for 7 consecutive days [20]. Co-administration of clarithromycin increased mean C max and AUC0–inf of vonoprazan by 1.6 and 1.4 times compared with when vonoprazan was administered alone. These data suggest that vonoprazan and clarithromycin mutually inhibit the metabolism of each other to a similar extent, probably via inhibition of CYP3A4. These data appear to offer contradicting evidence against the considerations based upon the in vitro drug interaction experiments (Sect. 3.1.3.2). Further clinical studies are required to study whether the co-administration of vonoprazan with other CYP3A substrate drugs alters their pharmacokinetics.
Oral administration of 10 mg vonoprazan once daily has been approved for the prevention of relapse after healing of aspirin- or non-steroidal anti-inflammatory drug-induced gastric or duodenal ulcer [22]. Pharmacokinetic studies were performed to investigate whether oral administration of low-dose aspirin (100 mg), loxoprofen sodium hydrate (60 mg), diclofenac sodium (25 mg), or meloxicam (10 mg) alters the pharmacokinetics of vonoprazan [20, 22]. These studies detected no changes in the pharmacokinetics of vonoprazan.
4 Pharmacodynamics
4.1 Mechanism of H+,K+-ATPase Inhibition by Vonoprazan
The discovery of the role of H+,K+-ATPase in the final step of gastric acid secretion fueled intensive research to find the ultimate anti-secretory agents. Historically, PPI was the first-in-class agent targeting this enzyme [8]. When H+,K+-ATPase is activated and the canalicular space is strongly acidic, PPI is converted non-catalytically to the active protonated sulfonamide, then forming a covalent disulfide bond with a cysteine residue on the H+,K+-ATPase leading to the inactivation of the enzyme [3]. Accordingly, PPIs inhibit mainly activated H+,K+-ATPase.
An alternative approach to inhibit H+,K+-ATPase activity is to reduce the availability of K+ for the enzyme. Because H+,K+-ATPase counter transports equal amounts of H+ and K+ to maintain an electrical balance through bi-directional ion transport, the availability of K+ for the enzyme is essential to maintain its activity [8]. P-CABs interfere with the binding of K+ to the enzyme in a competitive and reversible manner. Briefly, the H+,K+-ATPase localized at the apical membrane of the parietal cells is in E1 conformation and possesses a high-affinity H+ binding site on the cytoplasm side. Upon binding H+, the enzyme undergoes ATP-driven three-directional conformation changes to the phosphorylated E2 conformation (E2P), which possesses a high-affinity K+ binding site on the side of the extracellular canalicular space. Binding of K+ to E2P promotes dephosphorylation of the enzyme, which reverts to the E1 conformation and releases K+ into the cytoplasm. As the cycle is repeated, acid secretion continues [8]. Because P-CABs have higher pKa values than PPIs and are stable under low pH conditions, they are accumulated in the secretory canaliculi at higher concentrations than PPIs and act on the H+,K+-ATPase irrespective of its status of activity [24, 25].
Subsequent molecular studies on the H+,K+-ATPase have revealed that there is a single, high-affinity K+ binding site in the transmembrane helix [8]. P-CABs have been designed to bind at the vicinity of the K+ binding site. Differences in potency and duration of action among P-CABs may be associated with their affinity and/or dissociation constant for the binding sites on H+,K+-ATPase. For instance, vonoprazan has Ki values of 10 nmol/L at pH 7 [24] and 3 nmol/L at pH 6.5 [25], respectively. In addition, the IC50 for acid secretion is 17–19 nmol/L in porcine microsomes in in vitro studies [24]. In addition, the rate of dissociation of vonoprazan from isolated ATPase is slower than other P-CABs: the dissociation half-life of vonoprazan was 7.5 h in the presence of 20 mM KCL (near physiological concentration in stimulated gastric juice) as compared with <2 min for SCH28080 at 10 mH KCL [24]. The accumulation and clearance of vonoprazan from the gastric glands were studied by Matsukawa et al. using cultured rabbit gastric glands and compared with lansoprazole [29]. They demonstrated that the amounts of [14C]vonoprazan accumulated in the cultured rabbit gastric glands after a 2-h incubation were four and two times higher than those of lansoprazole at resting and forskolin-stimulated conditions, respectively. In addition, they studied the clearance of the two drugs from the glands by measuring the remaining radioactivity in the glands over a 24-h washout period. They found that the mean radioactivity of vonoprazan measured after washout was approximately seven and three times greater than those of lansoprazole at resting and forskolin-stimulated conditions, respectively.
4.1.1 Pharmacodynamics of Vonoprazan in Relation to Plasma Concentrations in Humans
Pharmacokinetic and pharmacodynamics profiles of vonoprazan were investigated in two phase I studies conducted in healthy subjects in Japan and the UK, who were given five different oral doses of vonoprazan or placebo once daily for 7 consecutive days [11]. Pharmacodynamic effects were measured by continuous intragastric pH monitoring. The onset of action measured by the increase in intragastric pH was rapid after oral dosing: the mean intragastric pH rose above 4 as early as 4 h after the first oral dose on day 1. Plasma concentrations of the drug reached the C max at 1.5–2.0 h post-dose, indicating that there was little time lag between the elevation of plasma concentrations and the increase in intragastric pH. The anti-secretory effect persisted throughout the 24-h interval before the next dose, in an apparently dose-dependent manner. With the recommended therapeutic dosage of 20 mg once daily, 24-h intragastric pH >4 holding time ratio (%HTR) on days 1 and 7 were 63 and 83 %, respectively, in Japanese patients and 63 and 85 % in UK patients. No population differences were observed in the dose–response relationship of vonoprazan. The night-time fall in intragastric pH was more attenuated in subjects given vonoprazan than in those given placebo, also in an apparently dose-dependent manner.
When the plasma concentration–effect relationship of the drug was evaluated based on plasma AUC0–tau and 24-h intragastric pH >4 %HTR, the concentration-effect curves obtained from Japanese and UK patients were almost superimposable, indicating that there were little population differences in the pharmacodynamics of vonoprazan between the two populations (Fig. 2). In addition, there was no attenuation in anti-secretory response based on the relationship between plasma AUC0–tau and 24-h intragastric pH >4 %HTR during administration of the drug over 7 consecutive days. Nevertheless, further studies are required to determine whether attenuation of an anti-secretory effect would emerge over a long period of use as observed for H2 receptor antagonists [30] (Fig. 2).

The relationships between plasma AUC0–tau of vonoprazan and 24-h in gastric pH >4 holding-time ratio (HTR) in healthy men (n = 60 and 48 for Japan and UK studies, respectively). The data are shown as the mean ± standard deviation. The figures were drawn based upon the data given in [11]
Assuming that the inhibition kinetics for H+,K+-ATPase obtained from in vitro studies using animal gastric vesicles [24, 25] can largely be extrapolated to humans, the mean plasma concentrations of unbound vonoprazan at C max after an oral dose of 20 mg is approximately 20–25 ng/mL (equivalent to 23–30 nmol/L), which would be comparable to the IC50 of H+,K+-ATPase. Given the competitive nature of inhibition, the achievement of almost complete suppression of acid secretion throughout the dosing interval of 24 h may be explained by accumulation of the drug in the secretory canaliculi of the parietal cells owing to its high pKa (9.06) as discussed in the previous Sect. (3.1.2) and slow dissociation from the enzyme [24]. Further studies are required to examine whether other demographic or pathophysiological factors are associated with the intra- or inter-individual variability in the anti-secretory effects of vonoprazan.
5 Safety and Tolerability
Because most of the clinical trials performed during the pre-approval period were reported only as abstracts [for review see 9, 13–19] and a summary of drug approval review [20], a detailed profile of adverse drug reactions cannot be evaluated at present. Although no studies have been undertaken or powered to assess this, vonoprazan appears to be well tolerated compared with PPIs. Among 2271 patients who received 10 or 20 mg of the drug for the treatment of acid-related disorders, 186 (8.2 %) developed adverse drug reactions including asymptomatic abnormalities in laboratory data. Among the reported clinical events, gastrointestinal symptoms of constipation (3.3 %), diarrhea (0.7 %), and flatulence (0.4 %) were the three most frequent symptoms. Because development of the prototype P-CAB SCH28080 was discontinued owing to hepatotoxicity [8], it is important to monitor the adverse drug reaction for newer P-CABs. Reversible abnormal liver function tests were observed in 0.2 % of the patients who received vonoprazan in pre-approval clinical studies: all events were reversible and most were associated with elevation of peak serum alanine transaminase or aspartate transaminase to <200 IU/L, while some were associated with elevation to >500 IU/L [20]. Because of the limited number of patients studied, a continuous post-marketing pharmacovigilance program for hepatotoxicity is required.
Another important safety issue of vonoprazan is hypochlorhydria-induced hypergastrinemia. Because gastrin has trophic effects on the epithelial cells of the stomach and intestine, sustained hypergastrinemia may have a role in the development of gastrointestinal malignancies. In preclinical toxicological studies, the administration of vonoprazan orally to male and female mice over 2 years was associated with the development of hyperplasia of neuroendocrine cells (enterochromaffin-like cells) or malignant carcinoid tumors at doses of 6 mg/kg/day or greater [20, 21]. A similar results were obtained with male and female rats at doses greater than 5 mg/kg/day or greater [20, 21]. A pharmacokinetic study performed with rats showed that plasma AUCs observed after the administration of vonoparazan associated with gastric tumors (i.e., 6 mg/kg) were largely comparable to those seen after a single oral dose of 40 mg in humans [20, 22]. It is uncertain whether the association between tumorigenesis and hypergastrinemia observed in rodents might be extrapolated into humans. A long-term administration of PPIs accompanying hypergastrinemia has not been demonstrated to increase the risk of developing carcinoid tumor owing probably to species differences in gastric physiology and enterochromaffin-like cells [31, 32]. Nevertheless, vigilant post-marketing surveillance should be undertaken until such theoretical concerns are negated by pharmacoepidemiological studies because vonoprazan was shown to elevate serum gastrin levels two to three times greater than PPIs [20–22].
6 Clinical Implications
Vonoprazan as monotherapy was approved for the treatment of gastroduodenal ulcer and reflux esophagitis, and the secondary prevention of low-dose aspirin or NSAID-induced gastric mucosal damages, and the prevention of relapse for reflux esophagitis. In addition, the drug was approved for the eradication of H. pylori as one of the first-line therapies in combination with clarithromycin and amoxicillin and as a second-line therapy in combination with amoxicillin and metronidazole [22].
Briefly, a phase III, randomized, double-blind clinical study was conducted in Japanese patients having endoscopically confirmed gastric ulcer [20, 21]. Two hundred and forty-four and 238 patients were allocated to receive either vonoprazan or lansoprazole at 20 and 30 mg once daily orally, respectively, for 8 weeks. Results demonstrated that the endoscopically confirmed healing was observed within 8 weeks in 93.5 % of patients receiving vonoprazan and 93.8 % of those receiving lansoprazole (95 % CI of the difference −4.8 to 4.2), indicating non-inferiority of vonoprazan as compared with lansoprazole, assuming the non-inferiority margin would be −8 %. In addition, another phase III, randomized, double-blind, clinical study was conducted for evaluating the efficacy of vonoprazan as compared with lansoprazole for the treatment of duodenal ulcer [20, 21]. One hundred and eighty-three and 185 Japanese patients were allocated to receive either vonoprazan or lansoprazole at 20 and 30 mg once daily, respectively, for 6 weeks. Results demonstrated that the endoscopically confirmed healing was observed within 6 weeks in 95.5 % of patients receiving vonoprazan and in 98.3 % of those received lansoprazole (95 % CI of the difference −6.4 to 0.7). Non-inferiority of vonoprazan as compared with lansoprazole failed to be confirmed, assuming the non-inferiority margin would be −6 %.
A phase III, randomized, double-blind, multicenter study was conducted for evaluating the efficacy and safety of vonoprazan (20 mg once daily) as compared with lansoprazole (30 mg once daily) in Japanese patients with endoscopically diagnosed reflux esophagitis [16, 20, 21]. Two hundred and seven and 202 patients were allocated to vonoprazan and lansoprazole, respectively, and the primary endpoint, endoscopically confirmed healing of esophagitis, was assessed at 2, 4, and 8 weeks after the beginning of the treatment. Results demonstrated that the healing rates by 8 weeks for vonoprazan and lansoprazole were 99.0 and 95.5 % (95 % CI of the difference 0.3–6.7), respectively, indicating the non-inferiority of vonoprazan as compared with lansoprazole, assuming the non-inferiority margin would be −6 % and superiority of vonoprazan.
As for the efficacy of vonoprazan for the eradication of H. pylori, a phase III, randomized, double-blind, multicenter study was performed in Japanese patients with active H. pylori infection confirmed by [13C]-urea breath test and endoscopically confirmed gastroduodenal ulcer scars [15, 20–22]. Three hundred and twenty-nine patients and 321 patients were allocated to receive either vonoprazan (20 mg twice daily) or lansoprazole (30 mg twice daily) with amoxicillin (750 mg twice daily) and clarithromycin (200 mg or 400 mg twice daily) for 7 days. Eradication of H. pylori was assessed by the urea breath test 4 weeks after the completion of the eradication therapy. Results demonstrated that the eradication rates for vonoprazan and lansoprazole were 92.6 and 75.9 %, respectively (95 % CI for the difference 11.2–22.1), indicating the non-inferiority of vonoprazan as compared with lansoprazole assuming the non-inferiority margin of −10 % and superiority of vonoprazan to lansoprazole. A phase III, randomized, double-blind study was conducted for evaluating the efficacy of vonoprazan (either 10 or 20 mg once daily orally) as compared with lansoprazole (15 mg once daily orally) for the secondary prevention of peptic ulcers in Japanese patients who needed a low-dose aspirin (81–324 mg daily) therapy for prevention of thromboembolic diseases despite having endoscopically confirmed healed peptic ulcers [14, 20]. Two hundred and two patients and 202 patients were allocated to low and high doses of vonoparazan and 217 patients were allocated to lansoprazole, and the clinical course was monitored for 24 weeks. Results showed that the relapsing rates of peptic ulcer in those given the low and high doses of vonoprazan (0.5 and 1.5 %, respectively) appeared lower than those given lansoprazole (2.8 %) and the 95 % CI for the differences between the low-dose vonoprazan vs. lansoprazole and the high-dose vonoprazan vs. lansoprazole were (−4.7 to 0.1) and (−4.1 to 1.5), respectively. As a result, the low and high doses of vonoprazan was considered non-inferior to lansoprazole assuming the non-inferiority margin of 8.7 %. Based upon these data, 10 mg vonoprazan was approved for clinical use. Another phase III study was conducted with a similar protocol for evaluating the efficacy of vonoprazan as compared with lansoprazole for the secondary prevention of gastroduodenal ulcers in Japanese patients who needed non-steroidal anti-inflammatory drugs for pain control of rheumatoid arthritis, osteoarthritis, and other orthopedic diseases despite having endoscopically confirmed healed peptic ulcers [13, 20–22]. Two hundred and eighteen and 212 patients were allocated to low and high doses (10 and 20 mg, respectively) of vonoparazan and 210 patients were allocated to lansoprazole, and the clinical course was monitored for 24 weeks. Results showed that the relapsing rates of peptic ulcer in those given the low and high doses of vonoprazan (3.3 and 3.4 %, respectively) appeared lower than that in those given lansoprazole (5.5 %). In addition, the 95 % CI for the difference in relapsing rate between low-dose vonoprazan vs. lansoprazole was −6.2 to 1.8 and for high-dose vonoprazan vs. lansoprazole was −6.1 to 2.0. As a result, the low and high doses of vonoprazan were considered non-inferior to lansoprazole assuming the non-inferiority margin of 8.3 %.
Collectively, the efficacy of vonoprazan seems largely non-inferior to lansoprazole in various clinical indications. Vonoprazan may have some advantages over PPIs in terms of the pharmacokinetic profile: more rapid onset of action, more powerful acid suppression particularly during night-time, and no influence of the CYP2C19 polymorphism on drug exposure. Regarding the pharmacodynamics profile, vonoprazan may be preferred to PPIs as maintenance therapy for reflux esophagitis and for eradication of H. pylori because of its stronger anti-secretory effect compared with PPIs [24].
References
Soll AH, Achord JL, Bozymski G, Brooks S, Lanza F, Lyon D, Meyer G, Reinus J, Schuster M, Achord J, Ofman J, Glassman P, Laine L, Tytgat G, Walsh JH, Graham DY, Peterson WL. Medical treatment of peptic ulcer disease: practice guidelines. JAMA. 1996;275:622–9.
Axon A. Helicobacter pylori and public health. Helicobacter. 2014;19(Suppl 1):68–73.
Fock KM, Ang TL, Bee LC, Lee EJD. Proton pump inhibitors. Clin Pharmacokinet. 2008;47:1–6.
Katz PO, Hatlebakk JG, Gstell DO. Gastric acidity and acid breakthrough with twice-daily omeprazole or lansoprazole. Aliment Pharmacol Ther. 2000;14:709–14.
Shaw MJ, Crawley JA. Improving health-related quality of life in gastro-oesophageal reflux disease. Drugs. 2003;63:2307–16.
Cheng HC, Sheu BS. Intravenous proton pump inhibitors in the management for peptic ulcer bleeding: clinical benefits and limits. World J Gastrointest Endosc. 2011;3:49–56.
Furuta T, Sugimoto M, Shirai N. Individualized therapy for gastroesophageal reflux disease: potential impact of pharmacogenetic testing based on CYP2C19. Mol Diagn Ther. 2012;16:223–34.
Parsons ME, Keeling DJ. Novel approaches to the pharmacological blockade of gastric acid secretion. Expert Opin Investig Drugs. 2005;14:411–21.
Chung IS, Choi MG, Park SH, et al. Revaprazan (Revanex®), a novel acid pump antagonist, for duodenal ulcer: results of a double-blind, randomized, parallel, multi-center phase III clinical trial. Korean J Gastrointest Endosc. 2005;31(1):17–24.
Chang R, Chung IS, Park SH, et al. Phase III clinical trial of revaprazan (Revanex®) for gastric ulcer. Korean J Gastrointest Endosc. 2007;34(6):312–9.
Jenkins H, Sakurai Y, Nishimura A, Okamoto H, Hibberd M, Jenkins R, Yoneyama T, Ashida K, Ogama Y, Warrington S. Randomised clinical trial: safety, tolerability, pharmacokinetics and pharmacodynamics of repeated doses of TAK-438 (vonoprazan), a novel potassium-competitive acid blocker, in healthy male subjects. Aliment Pharmacol Ther. 2015;41:636–48.
Umegaki E, Iwakiri K, Hiramatsu N, et al. A phase 3, randomized, double-blind, multicenter study to evaluate the efficacy and safety of TAK-438 (10 mg or 20 mg once-daily) compared to lansoprazole (15 mg once-daily) in a 24-week maintenance treatment for healed erosive esophagitis. Gastroenterology. 2014;146:S-738.
Mizokami Y, Ashida K, Soen S, et al. TAK-438 versus lansoprazole 15 mg for secondary prevention of peptic ulcers associated with non-steroidal anti-inflammatory drug (NSAID) therapy: results of a phase 3 trial. Gastroenterology. 2014;146:S-739.
Kawai T, Ashida K, Mizokami Y, et al. TAK-438 versus lansoprazole 15 mg for secondary prevention of peptic ulcers associated with low-dose aspirin therapy: results of a phase 3 trial. Gastroenterology. 2014;146:S-739.
Murakami K, Sakurai Y, Shiino M, et al. A phase 3, double-blind study of a triple therapy with TAK-438, amoxicillin, and clarithromycin as first line eradication of H. pylori and a triple therapy with TAK-438, amoxicillin, and metronidazole as second line eradication of H. pylori. Gastroenterology. 2014;146:S-740.
Iwakiri K, Umegaki E, Hiramatsu N, et al. A phase 3, randomized, double-blind, multicenter study to evaluate the efficacy and safety of TAK-438 (20 mg once-daily) compared to lansoprazole (30 mg once-daily) in patients with erosive esophagitis. Gastroenterology. 2014;146:S-741.
Sakurai Y, Mori Y, Okamoto H, et al. A crossover study to evaluate the acid-inhibitory effect of a newly developed potassium-competitive acid blocker, vonoprazan, compared with esomeprazole or rabeprazole. Gastroenterology. 2015;148:S-620.
Mizokami Y, Oda K, Saito K, et al. A phase 3, lansoprazole-controlled, long-term extension study to evaluate the safety and efficacy of vonoprazan (10 mg or 20 mg) for prevention of recurrent peptic ulcers during long-term therapy with non-steroidal anti-inflammatory drug (NSAID). Gastroenterology. 2015;148:S-88–S-89.
Garnock-Jones KP. Vonoprazan: first global approval. Drugs. 2015;75:439–43.
Drug approval review for vonoprazan fumarate (in Japanese), the Pharmaceuticals and Medical Devices Agency of Japan. 2014. https://www.pmda.go.jp/. Accessed 28 May 2015.
The interview form for vonoprazan fumarate (Takecab® tablets 10 mg and 20 mg) ver. 3, (in Japanese). Takeda Pharmaceutical Company Limited. 2015. http://www.takeda.co.jp/. Accessed 28 May 2015.
Prescribing information of Takecab® tablets 10 mg and 20 mg ver. 2 (in Japanese). 2015. http://www.takeda.co.jp/. Accessed 28 May 2015.
Arikawa Y, Nishida H, Kurasawa O, Hasuoka A, Hirase K, Inatomi N, Hori Y, Matsukawa J, Imanishi A, Kondo M, Tarui N, Hamada T, Takagi T, Takeuchi T, Kajino M. Discovery of a novel pyrrole derivative 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine fumarate (TAK-438) as a potassium-competitive acid blocker (P-CAB). J Med Chem. 2012;55:4446–56.
Shin JM, Inatomi N, Munson K, Strugatsky D, Tokhtaeva E, Vagin O, Sachs G. Characterization of a novel potassium-competitive acid blocker of the gastric H, K-ATPase, 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine monofumarate (TAK-438). J Pharmacol Exp Ther. 2011;339:412–20.
Hori Y, Imanishi A, Matsukawa J, Tsukimi Y, Nishida H, Arikawa Y, Hirase K, Kajino M, Inatomi N. 1-[5-(2-Fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine monofumarate (TAK-438), a novel and potent potassium-competitive acid blocker for the treatment of acid-related diseases. J Pharmacol Exp Ther. 2010;335:231–8.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry, drug interaction studies: study design, data analysis, implications for dosing, and labeling recommendations. 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed 26 Aug 2015.
European Medicine Agency (EMA), Committee for Human Medicinal Products (CHMP). Guideline on the investigation of drug interactions. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. Accessed 26 Aug 2015.
Ogawa R, Echizen H. Drug-drug interaction profiles of proton pump inhibitors. Clin Pharmacokinet. 2010;49:509–33.
Matsukawa J, Hori Y, Nishida H, Kajino M, Inatomi N. A comparative study on the modes of action of TAK-438, a novel potassium-competitive acid blocker, and lansoprazole in primary cultured rabbit gastric glands. Biochem Pharmacol. 2011;81:1145–51.
Sandvik AK, Brenna E, Waldum HL. Review article: the pharmacological inhibition of gastric acid secretion-tolerance and rebound. Aliment Pharmacol Ther. 1997;11:1013–8.
Freston JW. Omeprazole, hypergastrinemia, and gastric carcinoid tumors. Ann Intern Med. 1994;121:232–3.
Klinkenberg-Knol EC, et al. Long-term omeprazole treatment in resistant gastroesophageal reflux disease: efficacy, safety, and influence on gastric mucosa. Gastroenterology. 2000;118:661–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest regarding the present article.
Rights and permissions
About this article

Cite this article
Echizen, H. The First-in-Class Potassium-Competitive Acid Blocker, Vonoprazan Fumarate: Pharmacokinetic and Pharmacodynamic Considerations. Clin Pharmacokinet 55, 409–418 (2016). https://doi.org/10.1007/s40262-015-0326-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0326-7