
Abstract
Background and Objective
Metformin is contraindicated in patients with renal impairment; however, there is poor adherence to current dosing guidelines. In addition, the pharmacokinetics of metformin in patients with significant renal impairment are not well described. The aims of this study were to investigate factors influencing the pharmacokinetic variability, including variant transporters, between healthy subjects and patients with type 2 diabetes mellitus (T2DM) and to simulate doses of metformin at varying stages of renal function.
Methods
Plasma concentrations of metformin were pooled from three studies: patients with T2DM (study A; n = 120), healthy Caucasian subjects (study B; n = 16) and healthy Malaysian subjects (study C; n = 169). A population pharmacokinetic model of metformin was developed using NONMEM® version VI for both the immediate-release (IR) formulation and the extended-release (XR) formulation of metformin. Total body weight (TBW), lean body weight (LBW), creatinine clearance (CLCR; estimated using TBW and LBW) and 57 single-nucleotide polymorphisms (SNPs) of metformin transporters (OCT1, OCT2, OCT3, MATE1 and PMAT) were investigated as potential covariates. A nonparametric bootstrap (n = 1,000) was used to evaluate the final model. This model was used to simulate 1,000 concentration–time profiles for doses of metformin at each stage of renal impairment to ensure metformin concentrations do not exceed 5 mg/l, the proposed upper limit.
Results
Creatinine clearance and TBW were clinically and statistically significant covariates with the apparent clearance and volume of distribution of metformin, respectively. None of the 57 SNPs in transporters of metformin were significant covariates. In contrast to previous studies, there was no effect on the pharmacokinetics of metformin in patients carrying the reduced function OCT1 allele (R61C, G401S, 420del or G465R). Dosing simulations revealed that the maximum daily doses in relation to creatinine clearance to prescribe to patients are 500 mg (15 ml/min), 1,000 mg (30 ml/min), 2,000 mg (60 ml/min) and 3,000 mg (120 ml/min), for both the IR and XR formulations.
Conclusion
The population model enabled doses of metformin to be simulated for each stage of renal function, to ensure the concentrations of metformin do not exceed 5 mg/l. However, the plasma concentrations of metformin at these dosage levels are still quite variable and monitoring metformin concentrations may be of value in individualising dosage. This study provides confirmatory data that metformin can be used, with appropriate dosage adjustment, in patients with renal impairment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Type 2 diabetes mellitus (T2DM) is a chronic metabolic condition affecting over 340 million people worldwide (World Heath Organisation, 2012 [1]). Metformin is the drug of choice for patients with T2DM. It improves control by decreasing the rate of gluconeogenesis and increasing glucose uptake in the tissues [2]. It does not induce hypoglycaemia or weight gain [3]. Renal impairment is a common secondary complication of T2DM. Therefore, since metformin is largely renally cleared, it is widely stated to be contraindicated in patients with renal impairment (creatinine clearance (CLCR) <60 ml/min, Product Label [4]) as accumulation of the drug may be associated with lactic acidosis [5].
Metformin is a highly ionised, water-soluble substance that is absorbed, distributed and eliminated by transporters [6]. The best-documented transporters are the organic cation transporters (OCTs), which are localised in the liver (OCT1 and OCT3), apical (OCT1) and basolateral membranes of the kidney (OCT2) and the skeletal muscle (OCT3). Other transporters of metformin include the multidrug and toxin extrusion 1 transporter (MATE1), which transports metformin into the bile and the urine, and the plasma membrane monoamine transporter (PMAT), which transports metformin into intestinal cells [6–8].
Several factors influence the pharmacokinetics of metformin [9] and potentially affect its efficacy, tolerability and safety. First, the absorption of metformin is incomplete and variable (25–75 %), and decreases slightly with increasing dose [10, 11]. Secondly, the renal clearance of metformin is approximately fourfold higher than the creatinine clearance [6, 10], indicating substantial active tubular secretion. The renal clearance and total clearance of metformin (CL/F) decrease with decreasing CLCR but there is considerable scatter around the lines of best fit [6], indicating that other factors, such as variant low-function transporters, also control the renal clearance and CL/F [12].
Up to the present, detailed pharmacokinetic studies of metformin have been described mainly in small numbers of either healthy subjects or patients with T2DM [11, 13, 14]. There were two large-scale studies conducted in patients with T2DM [9, 15]. One study investigated the effect of single-nucleotide polymorphisms (SNPs) of metformin transporters on the trough plasma concentrations of metformin in patients with T2DM (n = 159), but the effect of renal function was not investigated [15]. In contrast, a population pharmacokinetic model was developed for patients with T2DM (n = 105), but the possible influence of low function transporters was not investigated [9]. Furthermore, no recommendations on dosage were made, and the pharmacokinetics of the extended-release formulation (XR) of metformin were not reported in these studies.
The therapeutic range of plasma concentrations of metformin is unclear; however, concentrations above 5 mg/l are considered elevated [16, 17]. In the present study, we have developed a population pharmacokinetic model for metformin combining immediate-release (IR) and XR data from both T2DM patients and healthy subjects. Factors potentially influencing the pharmacokinetics, such as CLCR and body weight, as well as the influence of variant genes of transporters of metformin were investigated. Lastly, our findings enabled time courses of metformin concentration to be simulated over a range of doses and kidney function to determine the maximum dose of metformin that should be prescribed to patients with renal impairment.
2 Methods
2.1 Study A
Study A (n = 120) consists of new data from patients treated with metformin for T2DM (n = 98) and published data from patients with chronic kidney disease (n = 22) [17]. All patients were recruited from the Diabetes Clinic, and there were no restrictions on the daily dose of metformin, dosing regimen or formulation of metformin (immediate-release, IR; XR). Other medications and oral antihyperglycaemic agents were permitted. Demographic characteristics and data on medical comorbidities and concurrent medications were collected. This study was approved by the Human Research Ethics Committee at St Vincent’s Hospital and University of New South Wales, Sydney (08209/SVH08/035), and was registered with the Australian New Zealand Clinical Trials Registry (ACTRN12611000908932).
In study A, a sparse sampling method was employed whereby one to six blood samples were collected from the patients. Patients were required to report times of their metformin doses over the preceding 2 days, dose strength, frequency and compliance to their medications. The timing of the blood samples was selected to obtain peak concentrations of metformin (C max; IR, 2–4 h post-dose; XR, 4–6 h post-dose) and trough concentrations of metformin (C min; IR, 10–12 h post-dose; XR, 20–24 h post-dose). Glycosylated haemoglobin (HbA1C) and lactate concentrations were determined on the first visit.
Selected patients (n = 16) were involved in an intensive sampling sub-study. Serial blood samples were collected over the dosing interval (IR, 12 h; XR, 24 h). For patients taking metformin IR, serial venous blood samples were collected at 0 (pre-dose), 0.5, 1, 2, 3, 4, 6, 8, 10 and 12 h after metformin administration. For patients taking metformin XR, blood samples were collected at 0, 2, 4, 6, 12, 16, 20 and 24 h after a metformin dose.
Metformin concentrations in plasma were determined using a validated, reverse-phase high-performance liquid chromatography assay utilising a Zorbax Cyano column (150 × 4.6 mm, 5 µm particles) with the temperature set to 40 °C [17]. The mobile phase consisted of methanol:acetonitrile:phosphate buffer (20:60:20, v:v; pH adjusted to 7.0 with acetic acid). Quantification was achieved using UV detection (236 nm) and peak height ratios of six calibration standards (0.05–10 mg/l) with two quality control samples (QCs; 1.0 and 3.0 mg/l). The lower limit of quantification (LOQ) was 0.05 mg/l. The average between-day accuracy (% difference from nominal value) of the QCs was −0.4 % with a precision (measured as the coefficient of variation) of 0.8 %. The standard curves were linear (R 2 ≥ 0.997) with peak purities ≥98 % by spectral examination.
2.1.1 Genotyping
Patients were genotyped for a total of 59 SNPs in OCT1, OCT2, OCT3, MATE1 and PMAT transporters (Supplementary Table II and III). The DNA was extracted using the phenol–chloroform method and all patients were genotyped using an oligo-ligation assay (iPLEX Gold™) with the MassARRAY platform (Sequenom) in duplicate. SNP analysis was performed with the software Typer 4.0.3. Patients were excluded from genetic analysis if over 10 % of genetic information was missing (>6 genotypes) or if genetic information was conflicting in duplicate runs. Similarly, SNPs were excluded if there was over 10 % of patients with a missing genotype (>12 patients).
2.2 Studies B and C
Studies B and C consist of healthy subject data from two different populations that were supplied to us. Mean data from study B (n = 16) on healthy Caucasian subjects has been published [11]. This study was designed as a multiple-dose, five-regimen, two-sequence clinical study (see Supplementary Table I) [11]. Study C consists of unpublished data from healthy Malaysian subjects. This was a single-dose bioequivalence study approved by the Joint Penang Independent Ethics Committee (04-0014a 04-0014b, 05-0072, 06-0133, 06-0155, 07-0245, 08-0274, 08-0280, 08-0282). Subjects from studies B and C were not genotyped for SNPs in metformin transporters.
2.3 Pharmacokinetics of Metformin
Data from the three studies were analysed by non-linear mixed effect modelling (NONMEM® VI) [19] with first order conditional estimation method with interaction (FOCE-I). Model selection was informed by using the objective function value (OFV, −2log likelihood) [20] and visual inspection of diagnostic plots. A difference of ≥3.84 in OFV was considered statistically significant (p < 0.05, χ2 distribution).
2.3.1 Structural Model
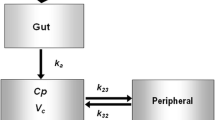
Several structural pharmacokinetics models, including one- and two-compartment models, with and without lag times, were tested during model development. The final structural model was parameterised in terms of CL/F, apparent central and peripheral volumes of distribution (V 1/F and V 2/F), apparent inter-compartmental clearance (Q/F), a first order absorption (k a) for the IR formulation and a zero-order input for the XR formulation. A lag time for absorption (ALAG) was used for the IR formulation.
The structural model was first built using the data from study B, which contained rich data sets of healthy subjects at steady state on metformin IR (2,000 mg/day) and XR doses (500, 1,000, 1,500 and 2,000 mg daily). Inclusion of oral availability (F) at each dose of metformin was investigated. This model was then used as a basis for providing initial estimates for studies A, B and C. All key model building steps were undertaken once all data had been included in the final data set to ensure appropriate model building decisions were made.
Inter-individual variability (IIV) is the unexplained random variability observed between subjects and introduced sequentially for each pharmacokinetic parameter. The inter-occasion variability (IOV) is the unexplained random variability observed within the individual on different occasions; an occasion was defined as observations following a dose of metformin. Population parameter variability (PPV) was modelled using both IIV and IOV and was modelled as an exponential function assuming log-normal distribution. The PPV was introduced in a stepwise manner for CL/F and V 1/F and was included in the final structural model if there was a statistically significant drop in OFV (p < 0.05). A mixed (additive and proportional) error model was used to describe the residual error in the data. A separate residual error model was evaluated for each of the three studies [21].
2.3.2 Covariate Model
The influence of demographic and biochemical covariates was assessed by visual inspection of the covariate and IIV plots. Potential covariates were incorporated sequentially into the model in order of significance, whereby the forward inclusion of the covariate was based on a significant drop in OFV, biological plausibility, a drop in the IIV and the observed change in the parameter values as a result of the covariate inclusion being clinically significant (>20 % change in parameter value across the range of the covariate). The covariates analysed were total body weight (TBW), lean body weight (LBW) [22, 23] and age. CLCR was also investigated as a potential covariate and was calculated using the Cockcroft-Gault equation [24], with either TBW or LBW. These covariates were normalised to the median population value of 70 kg for body weight and 6 l/h for CLCR to enable faster computation by NONMEM®.
The effect of SNPs on the pharmacokinetics was examined based on biological plausibility. Since most of the variant transporters investigated were localised in the kidneys, the effect of the SNPs on CL/F was evaluated statistically. All continuous variables were assessed via linear regression and all categorical variables were assessed using box and whisker plots. Information on the use of concomitant medications and variant genes of the transporters were only examined for study A. The backward elimination of covariates was used to confirm covariate selection, whereby an increase in OFV >6.63 (p < 0.01) was required.
2.3.3 Model Evaluation
The goodness of fit was evaluated by visual inspection of diagnostic scatter plots, including the observed and predicted concentrations versus time, conditional weighted residual versus predicted concentrations and normalised prediction distribution errors (NPDEs). The conditional weighted residual is the preferred diagnostic tool for population models developed using the FOCE-I method [25]. NPDEs account for the full predictive distribution of each observation and are useful for evaluating simulated data with the original data [26]. A nonparametric bootstrap method [27] (n = 1,000) was used to study the uncertainty of all pharmacokinetic parameter estimates in the final base model. From the bootstrap empirical posterior distribution, we were able to obtain the 95 % confidence interval (2.5–97.5 %) for the parameters, as described previously [27]. The final model performance was also examined by visual predictive checks, by comparing the 10th, 50th and 90th percentiles of the observed concentrations and simulations of concentration–time profiles (1,000 replicates) from the final model, when the dose was normalised to 1,000 mg. The bootstrap was performed using Wings for NONMEM® (http://wfn.sourceforge.net/).
2.3.4 Dosing Simulations
Dosing simulations were used to evaluate a range of metformin doses (500, 1,000, 2,000 and 3,000 mg) at varying levels of renal function (CLCR; 15, 30, 60 and 120 ml/min) to ensure that the 95th percentile of metformin C max stayed below 5 mg/l. Using the final population pharmacokinetic model, metformin concentrations were simulated at steady state for metformin IR and XR. The impact of between-occasion variability (BOV) on the simulations was tested. Each stochastic simulation generated 1,000 concentration–time profiles for all subjects per dose and renal function group.
2.4 Statistical Methods
Statistical analyses were performed using SPSS (version 20.0; SPSS, Chicago, IL, USA). The data are presented as median and range. The genotype frequencies were detected by the Hardy-Weinberg equilibrium test [28]. The effect of diuretics on metformin pharmacokinetics was assessed using an unpaired t test. For three genotype groups (WT, wild-type; WT/V, heterozygous variant; V/V, homozygous variant) then the effect was assessed using a one-way analysis of variance (ANOVA). The unpaired t test was used to investigate the effect of SNPs if there were only two genotype groups (WT, WT/V). The average plasma concentration of metformin at steady state (C av,ss) was used to investigate correlations with HbA1C and lactate concentrations. A p value of <0.05 was considered statistically significant.
3 Results
3.1 Patients and Healthy Subjects
The overall data set was derived from a total population (n = 304) from three different studies. Demographic information for all subjects is summarised in Table 1. In general, patients with T2DM were older (p < 0.001) and had lower renal function than the healthy population (p < 0.001).
In study A, there were 52 patients (43 %) with renal impairment (CLCR <60 ml/min), including 13 patients (11 %) with chronic kidney disease (CLCR values below 30 ml/min). Additionally, 33 patients (28 %) were taking at least one diuretic [thiazide, furosemide (frusemide) or potassium-sparing diuretic]. The median daily dose of metformin was 1,500 mg (range 250–3,000 mg). There was variable glycaemic control in the patients with T2DM and the daily dose of metformin was higher in patients with poor glycaemic control (HbA1C >7.5%; p < 0.05). About half of the patients were taking metformin with another oral antihyperglycaemic agent, mostly sulfonylureas. Most patients with T2DM had normal plasma concentrations of lactate but there were 13 patients with lactate concentrations above the reference range (>2.7 mmol/l).
3.2 Pharmacokinetics
A total of 4,895 metformin plasma concentrations were used for population pharmacokinetic analyses. The metformin concentration–time data were best described by a two-compartment model with first order absorption for the IR and zero-order absorption for XR formulations. Inter-individual variability (IIV) was included in all parameters but could not be established for the rate constant of absorption (k a) for IR (set at 0.35 h−1) and the duration of infusion for XR (set at 9.55 h). The relative oral availability (F) of metformin decreased with increasing XR doses, with the mean relative F decreasing from 1.26 at 500 mg, to the reference value of 1 at 2,000 mg daily (Table 2). The effect of F on different IR doses was tested; however, there were no consistent trends with increasing dose. Therefore, the value of F was fixed to 1 for all IR doses. Models to evaluate a continuous function of oral availability against dose were evaluated during model development as described previously [29]. However, these did not appear to describe the data as well as the discrete dose oral availability model based on objective function and diagnostic plots.
The addition of a separate residual error for each of the three studies significantly improved the model (ΔOFV = −1,038). The correlations between pharmacokinetic parameters (CL/F, V 1/F, Q/F and V 2/F) were calculated to be below 0.95 (Table 2). The IOV on CL/F and V 1/F were 9.5 and 17.8 %, respectively, and their inclusion significantly improved the model (ΔOFV = −176).
A nonparametric bootstrap (n = 1,000 replicates), together with the lack of correlation between the kinetic parameters, showed that the final model was robust (Table 2). Figure 1 displays the goodness-of-fit and diagnostic plots for this model. The conditional weighted residual graph showed no apparent visual or statistical bias for the prediction. Individual metformin concentrations were over-predicted in five patients (Fig. 1d). Since subsequent observations collected from these patients were well predicted by the model, this over-prediction may have been due to poor patient compliance or to lower absorption. However, the evaluation of models to account for possible altered compliance was not successful. The visual predictive check showed good agreement between the 10th and 90th percentiles of observed data and simulated data (Fig. 2).

Goodness-of-fit plots and diagnostic plots of the population pharmacokinetic model of metformin. a Conditional weighted residuals vs. population predicated concentrations, b normalised prediction distribution errors (NPDE) plots, c observed concentrations vs. population predicted concentrations and d individual predicted concentrations. Open circles observed data points, solid line line of identity

Visual predictive check (VPC) of the final model of metformin describing the pharmacokinetics of patients with type 2 diabetes mellitus and healthy subjects taking either the immediate-release or extended-release formulation. VPCs in a and b are not prediction-corrected, whilst c and d are prediction-corrected VPCs. The observed data (red circles) were plotted with the 10th, 50th and 90th percentiles (red lines) and the 10th, 50th and 90th percentiles of the predictions (black lines) were plotted with the 95 % confidence interval (shaded grey area)
The inclusion of CLCR (calculated using LBW) on metformin CL/F, and TBW on V 1/F, reduced the IIV from 44 to 34 % and 62 to 56 %, respectively. Subsequent removal of either one of these covariates produced a poorer fit to the model. The age of the patients was not a significant covariate. Additionally, the use of diuretics did not influence the clearance of metformin (p = 0.104). The estimates of eta shrinkage and epsilon shrinkage were low (<20 %; Table 2). The final equations for CL/F and V 1/F were (Eq. 1 and 2):
where θ CL and \( \theta_{{V_{ 1} }} \) are the mean population value for CL/F and V 1/F, respectively. PPVCL and PPVV1 are the sum of the IIV and IOV for either CL/F or V 1/F. The covariates, CLCR and TBW, were normalised to the median population value.
A non-renal component in the metformin CL/F did not improve the model and was therefore not included. Furthermore, in the regression CL/F versus CLCR, the intercept was not significantly different from 0 and therefore metformin CL/F decreased proportionally with decreasing CLCR. This indicates that the non-renal clearance component of CL/F is not significant.
Overall, the median apparent clearance of metformin for studies A, B and C were 760 ml/min (range 156–2,307 ml/min), 1,201 ml/min (800–1,762 ml/min) and 1,066 ml/min (564–3,048 ml/min). The median CL/F-to-CLCR ratio was 12.3 (range 5.6–42.5). Metformin C av,ss for studies A and B was 1.28 mg/l (range 0.2–7.7 mg/l) and 0.9 (0.6–1.1 mg/l), respectively. A patient with end-stage kidney disease (CLCR = 15 ml/min) had the highest C av,ss (7.7 mg/l) and was on high doses of 2,000 mg metformin daily.
3.3 Effects of Variant Transporters on Metformin Pharmacokinetics
Two SNPs (OCT3, rs62440430; MATE1, rs2252281) and 17 patients were excluded from genetic analyses of metformin pharmacokinetics because of a high rate of missing genotype information (>10 %) and conflicting results in duplicates. Therefore, the effects of 57 SNPs in 103 patients were investigated (Table 3). All SNPs were in Hardy-Weinberg equilibrium (p > 0.01). None of the SNPs investigated had a statistically significant effect on metformin CL/F. In particular, of the 103 patients, 66 had one low activity variant of the OCT1 gene (Arg61Cys, Gly401Ser, M420del or Gly465Arg), and 2 patients had two low activity variants of OCT1. The contrast with the reference group was not significant. Therefore, SNPs were not added as a covariate on the model.
3.4 Correlations between Metformin Concentrations and Effect
For patients with T2DM, there was no correlation between metformin C av,ss with either lactate concentrations or HbA1C (Supplementary Figure 1). If patients on metformin monotherapy were grouped separately, there was, however, a weak correlation between metformin C av,ss and HbA1C (R 2 = 0.187, p < 0.01). Surprisingly, the slope was positive whereby HbA1C increased with increasing C av,ss. However, this correlation was not found in patients taking metformin with other oral antihyperglycaemic agents. In addition, there were no correlations between metformin C av,ss and lactate concentrations in either metformin monotherapy or combination therapy (Supplementary Figure 1).
3.5 Simulations and Dose Recommendations
Since the concentration of metformin should not exceed 5 mg/l, this was used as the maximum concentration of metformin for dose simulations. BOV did not influence the concentrations of metformin and was therefore removed. Proposed IR doses were made for each of the levels of renal function, namely, at CLCR of 15, 30, 60 and 120 ml/min (Fig. 3). Dosing simulations revealed that to ensure the 95th percentile of metformin C max is below 5 mg/l, maximum IR and XR doses of 500, 1,000, 2,000 and 3,000 mg daily should be prescribed to patients with a respective CLCR of 15, 30, 60 and 120 ml/min. However, metformin concentrations were above 5 mg/l (5.4 mg/l, 95th percentile) for patients with CLCR of 60 ml/min at doses of 2,000 mg XR, but not at daily doses of 2,000 mg IR (5 mg/l). The maximum recommended dose of metformin XR is 2,000 mg daily, but patients with good renal function can be dosed 3,000 mg XR daily without exceeding metformin concentrations of 5 mg/l. At each level of CLCR, the predicted 95 % confidence range of metformin concentrations was considerable and indicated substantial inter-patient differences in CL/F, which was not explained by the covariates, CLCR and TBW.

Simulations of the plasma concentrations at maximum doses of metformin for patients with varying degrees of renal function. The 5th, 50th and 95th percentiles of the simulated concentrations are shown. The maximum daily dose of metformin immediate release (IR) or extended release (XR) that should be given to patients with creatinine clearances of 15, 30, 60 or 120 are 500, 1,000, 2,000 or 3,000 mg, respectively. The IR doses are reported as equal split doses
The elimination half-life was similar for both IR and XR formulations. However, they were longer for patients with renal impairment, whereby at CLCR of 15, 30 and 60 ml/min, the median half-lives were about 13, 8 and 4.5 h, respectively. At a CLCR of 120 ml/min, the half-life for IR was monophasic and was about 3 h. However, the half-life was biphasic for the XR formulation and was approximately 1.2 and 7 h for the initial and late phase.
4 Discussion
The pharmacokinetics of metformin for the IR and XR formulations were successfully described by developing a population model using a large data set from healthy subjects and patients with T2DM. Previous population pharmacokinetic models, built using data from patients with T2DM, have described metformin pharmacokinetics using a one-compartment model [9, 14]. A one-compartment model is sufficient for IR tablets as they are often administered every 8 h but the influence of the two-compartment model can be seen in the bi-exponential elimination phase in the 15 h following the absorption phase of the XR doses. Our two-compartment model is also more biologically plausible given the biphasic decline in the plasma concentrations of metformin after single doses [6]. It has been suggested that the slow uptake of metformin into red blood cells may be part of this second deep compartment [10, 30].
Similar to previous pharmacokinetic studies, CL/F was proportional to CLCR [6, 14]. CLCR was estimated using LBW, which is more appropriate for patients with T2DM because use of TBW overestimates CLCR, particularly in obese patients [22]. Considering that CL/F is larger than the renal clearance of metformin, largely because of incomplete absorption (low F), and furthermore that any non-renal clearance of metformin was not a significant covariate in our study, it is not surprising that CL/F should also be approximately proportional to CLCR.
In contrast to the results of Sambol et al. [31], we found that age was not a covariate for CL/F once CLCR has been added. It should be noted that including colinear covariates, such as age and CLCR, may result in biased parameter estimates [32]. The median CL/F for the healthy subjects was comparable to metformin CL/F reported in previous pharmacokinetic studies (933–1,317 ml/min) [9, 14]. In comparison, patients with T2DM had lower median CL/F of metformin (760 ml/min; study A) as they had lower median CLCR (67 ml/min). Our results indicate that CLCR is proportional to CL/F, which is consistent with the ‘intact nephron hypothesis’, whereby clearance by filtration is proportional to any clearance by secretion [33].
It has been previously reported that low functional alleles of OCT1 were associated with slightly higher plasma concentrations of metformin and lower mean CL/F than the reference group [12]. However, we found no significant effect of SNPs of OCT1, OCT2, OCT3, MATE1 or PMAT genes on the CL/F of metformin. Consistent with our results, Christensen et al. [15] found that trough plasma concentrations of metformin in 65 heterozygotes carrying any one of the four low-function alleles of OCT1 were not different from 72 patients in the reference group. However, Christensen et al. [15] reported an overall trend to lower trough plasma concentrations in ten patients with two low-function alleles. In contrast, only 2 of 103 patients in our study had two low-function alleles of OCT1 (Supplementary Table II). This marked difference in the occurrence of haplotypes requires further study. The limited number of patients with more than one low function allele of OCT1 in our study prevented any statistical conclusions on the influence of haplotypes in our study.
In our study, there were no restrictions on the dose or formulation of metformin and the large pharmacokinetic variability between patients was captured in this model. Apart from compliance, CL/F is related to the oral availability, F. The inter-individual variability in CL/F was 34 %, which would be reduced if F could be determined for each patient. Conducting cross-over studies between oral and intravenous dosage would enable this variability to be characterised but would be difficult to conduct in routine clinical practice, as was the case in our study. In addition, CL/F and F appear to be dependent upon transporters but, at this stage, reasons for the inter-patient variability can only be speculative. One possibility is the marked variation in the levels of expression: the approximately 100-fold variation in the expression of OCT1 and the 27-fold variation of OCT3 in the liver [34] and the 100-fold variation of OCT2 in the kidney [35]. However, since SNPs in metformin transporter genes did not have an observable effect on the CL/F of metformin, we conclude that, at this time, testing for the patient’s genotype is not useful when modifying dosage.
We have suggested previously that the C av,ss of metformin over a dosage interval may be a better indicator of the exposure of the body to metformin than the peak or trough concentrations [6]. However, correlations between C av,ss with either HbA1C or lactate concentrations were found to be either low or not significant. A few patients, including patients with chronic kidney disease, had lactate concentrations above the reference range (>2.7 mmol/l). However, as reported previously, repeated biochemical measures in these patients did not support a diagnosis of lactic acidosis and their treatment with metformin was continued [17].
Several reasons may be suggested for the weak correlation between HbA1C and the C av,ss of metformin. Firstly, patients with poor glycaemic control were prescribed higher doses of metformin than patients with better control of their T2DM. Since these patients were recruited from the specialist Diabetes Clinic, they represent more complex cases of T2DM with early signs of nephropathy. Therefore, CLCR was a useful covariate as patients from study A had lower CLCR than subjects from study B and C. Secondly, the baseline (pre-metformin) concentrations of HbA1C and lactate concentration with metformin treatment were not available and, therefore, the change in these parameters could not be determined. The decrease in HbA1C has been measured in a previous study together with trough concentrations of metformin [15] but a full pharmacokinetic analysis and examination of covariates was not attempted. Studies on the measurement of HbA1C before and during treatment of metformin together with examination of pharmacokinetic parameters may enable better pharmacokinetic–pharmacodynamic relationships to be established.
Prescribing metformin to patients with renal impairment remains a challenging issue for clinicians [36, 37]. The current guidelines are inconsistent, whereby metformin is either contraindicated in patients with CLCR values of 30 ml/min (Australian Medicines Handbook) [38] or 60 ml/min (Product Information [4]). However, metformin is frequently prescribed to patients with renal impairment. For example, 20–34 % of patients with T2DM with estimated GFR ≤60 ml/min were prescribed metformin in two studies [39, 40]. Similarly, 43 % of patients from study A had CLCR values <60 ml/min, including five patients who were monitored on low doses of metformin [17].
Knowledge of metformin pharmacokinetics in patients at varying stages of kidney disease is limited. Reports have been for either single-dose studies [31] or studies interpreting the pharmacokinetics based upon single concentrations of metformin from each patient [39, 41]. Our study incorporated patients with renal impairment [17]. Several of these patients had multiple samples of metformin collected on several occasions. Additionally, the population model was used to simulate doses in patients at various stages of kidney function. Using a conservative approach such that the 95th percentile of metformin C max did not exceed 5 mg/l, the maximum daily dose of metformin IR and XR that should be prescribed to a patient with a creatinine clearance of 15, 30, 60 and 120 ml/min are 500, 1,000, 2,000 and 3,000 mg, respectively. At these doses the median C av,ss of metformin was 1.5 mg/l (range 1.1–1.5 mg/l). Although the maximum recommended dose of metformin XR is 2,000 mg daily (Product Information [4]), the simulations showed that it is possible for patients with normal kidney function to be prescribed 3,000 mg XR daily, with the 95 % percentile of metformin C max slightly exceeding the upper limit (5.2 mg/l). It should be noted that there is considerable variability in the concentrations of metformin at each dose level, with a small number of patients outside the 5th and 95th percentile range. The plasma concentrations of metformin should be monitored in addition to adjusting initial target doses according to renal function.
5 Conclusion
In summary, a population pharmacokinetic model was developed for metformin for various dosing regimens, dose formulations and degrees of renal function. The simulations indicate that metformin can generally be used in patients with renal impairment while avoiding metformin concentrations above 5 mg/l. This present model provides a guide to dosing in patients taking metformin, particularly in patients with renal impairment.
References
World Health Organization. Diabetes fact sheet no. 312. 2012. http://www.who.int/mediacentre/factsheets/fs312/en/index.html. Accessed 19 Feb 2012
Hundal RS, Krssak M, Dufour S, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49(12):2063–9.
Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):854–65.
Diabex tablets. Carole Park, Australia: Alphapharm Pty Ltd; 2012
Bruijstens LA, van Luin M, Buscher-Jungerhans PM, et al. Reality of severe metformin-induced lactic acidosis in the absence of chronic renal impairment. Neth J Med. 2008;66(5):185–90.
Graham GG, Punt J, Arora M, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98.
Tzvetkov MV, Vormfelde SV, Balen D, et al. The effects of genetic polymorphisms in the organic cation transporters OCT1, OCT2, and OCT3 on the renal clearance of metformin. Clin Pharmacol Ther. 2009;86(3):299–306.
Zhou M, Xia L, Wang J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab Dispos. 2007;35(10):1956–62.
Bardin C, Nobecourt E, Larger E, et al. Population pharmacokinetics of metformin in obese and non-obese patients with type 2 diabetes mellitus. Eur J Clin Pharmacol. 2012;68(6):961–8.
Tucker GT, Casey C, Phillips PJ, et al. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. 1981;12(2):235–46.
Timmins P, Donahue S, Meeker J, et al. Steady-state pharmacokinetics of a novel extended-release metformin formulation. Clin Pharmacokinet. 2005;44(7):721–9.
Shu Y, Brown C, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83(2):273–80.
Sambol NC, Chiang J, O’Conner M, et al. Pharmacokinetics and pharmacodynamics of metformin in healthy subjects and patients with noninsulin-dependent diabetes mellitus. J Clin Pharmacol. 1996;36(11):1012–21.
Hong Y, Rohatagi S, Habtemariam B, et al. Population exposure-response modeling of metformin in patients with type 2 diabetes mellitus. J Clin Pharmacol. 2008;48(6):696–707.
Christensen MM, Brasch-Andersen C, Green H, et al. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharmacogenet Genomics. 2011;21(12):837–50.
Lalau JD, Lemaire-Hurtel AS, Lacroix C. Establishment of a database of metformin plasma concentrations and erythrocyte levels in normal and emergency situations. Clin Drug Investig. 2011;31(6):435–8.
Duong JK, Roberts DM, Furlong TJ, et al. Metformin therapy in patients with chronic kidney disease. Diabetes Obes Metab. 2012;15(10):963–5.
Zarghi A, Foroutan SM, Shafaati A, et al. Rapid determination of metformin in human plasma using ion-pair HPLC. J Pharm Biomed Anal. 2003;31(1):197–200.
Boeckman A, Sheiner A, Beal S. NONMEM 6. Ellicott City: GloboMax, ICON Development Solutions; 2007.
Wang Y. Derivation of various NONMEM estimation methods. J Pharmacokinet Pharmacodyn. 2007;34(5):575–93.
Cullberg M, Eriksson UG, Larsson M, et al. Population modelling of the effect of inogatran, at thrombin inhibitor, on ex vivo coagulation time (APTT) in healthy subjects and patients with coronary artery disease. Br J Clin Pharmacol. 2001;51(1):71–9.
Janmahasatian S, Duffull SB, Chagnac A, et al. Lean body mass normalizes the effect of obesity on renal function. Br J Clin Pharmacol. 2008;65(6):964–5.
Janmahasatian S, Duffull SB, Ash S, et al. Quantification of lean bodyweight. Clin Pharmacokinet. 2005;44(10):1051–65.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187–97.
Brendel K, Comets E, Laffont C, et al. Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res. 2006;23(9):2036–49.
Henderson AR. The bootstrap: a technique for data-driven statistics. Using computer-intensive analyses to explore experimental data. Clin Chim Acta. 2005;359(1–2):1–26.
Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy–Weinberg equilibrium. Am J Hum Genet. 2005;76(5):887–93.
Roos JF, Kirkpatrick CM, Tett SE, et al. Development of a sufficient design for estimation of fluconazole pharmacokinetics in people with HIV infection. Br J Clin Pharmacol. 2008;66(4):455–66.
Lalau JD, Lacroix C. Measurement of metformin concentration in erythrocytes: clinical implications. Diabetes Obes Metab. 2003;5(2):93–8.
Sambol NC, Chiang J, Lin ET, et al. Kidney function and age are both predictors of pharmacokinetics of metformin. J Clin Pharmacol. 1995;35(11):1094–102.
Bonate PL. The effect of collinearity on parameter estimates in nonlinear mixed effect models. Pharm Res. 1999;16(5):709–17.
Bricker NS, Morrin PA, Kime SW Jr. The pathologic physiology of chronic Bright’s disease. An exposition of the “intact nephron hypothesis”. Am J Med. 1960;28:77–98.
Nies AT, Koepsell H, Winter S, et al. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology. 2009;50(4):1227–40.
Aoki M, Terada T, Kajiwara M, et al. Kidney-specific expression of human organic cation transporter 2 (OCT2/SLC22A2) is regulated by DNA methylation. Am J Physiol Ren Physiol. 2008;295(1):F165–70.
Kamber N, Davis WA, Bruce DG, et al. Metformin and lactic acidosis in an Australian community setting: the Fremantle Diabetes Study. Med J Aust. 2008;188(8):446–9.
Nye HJ, Herrington WG. Metformin: the safest hypoglycaemic agent in chronic kidney disease? Nephron Clin Pract. 2011;118(4):c380–3.
Rossi S. Australian medicines handbook 2011. Adelaide: Australian Medicines Handbook Pty Ltd; 2011.
Frid A, Sterner GN, Londahl M, et al. Novel assay of metformin levels in patients with type 2 diabetes and varying levels of renal function: clinical recommendations. Diabetes Care. 2010;33(6):1291–3.
Kamber N, Davis WA, Bruce DG, et al. Metformin and lactic acidosis in an Australian community setting: the Fremantle Diabetes Study. Med J Aust. 2008;188(8):446–9.
Briet C, Saraval-Gross M, Kajbaf F, et al. Erythrocyte metformin levels in patients with type 2 diabetes and varying severity of chronic kidney disease. Clin Kidney J. 2012;5(1):65–7.
Acknowledgements
The authors would like to thank Prof. Kathleen Giacomini for advice on the selection of SNPs in metformin transporters and Dr. Pavel Bitter for the analyses of SNPs. Funding for this study was provided by the NH&MRC Programme Grant 568612, Australian Research Council Grant LP 0990670, and St Vincent’s Clinic Foundation Sister Mary Bernice Research Grant.
Conflicts of interest
Peter Timmins is a salaried employee of Bristol-Myers Squibb, which is involved in the development and marketing of products containing metformin. Apart from the salary of Dr. Timmins, Bristol-Myers Squibb made no other payments in the support of this work. All other authors declared no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Duong, J.K., Kumar, S.S., Kirkpatrick, C.M. et al. Population Pharmacokinetics of Metformin in Healthy Subjects and Patients with Type 2 Diabetes Mellitus: Simulation of Doses According to Renal Function. Clin Pharmacokinet 52, 373–384 (2013). https://doi.org/10.1007/s40262-013-0046-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0046-9