
Abstract
New oral anticoagulants (OACs) that directly inhibit Factor Xa (FXa) or thrombin have been developed for the long-term prevention of thromboembolic disorders. These novel agents provide numerous benefits over older vitamin K antagonists (VKAs) due to major pharmacological differences. VKAs are economical and very well characterized, but have important limitations that can outweigh these advantages, such as slow onset of action, narrow therapeutic window and unpredictable anticoagulant effect. VKA-associated dietary precautions, monitoring and dosing adjustments to maintain international normalized ratio (INR) within therapeutic range, and bridging therapy, are inconvenient for patients, expensive, and may result in inappropriate use of VKA therapy. This may lead to increased bleeding risk or reduced anticoagulation and increased risk of thrombotic events. The new OACs have rapid onset of action, low potential for food and drug interactions, and predictable anticoagulant effect that removes the need for routine monitoring. FXa inhibitors, e.g. rivaroxaban and apixaban, are potent, oral direct inhibitors of prothrombinase-bound, clot-associated or free FXa. Both agents have a rapid onset of action, a wide therapeutic window, little or no interaction with food and other drugs, minimal inter-patient variability, and display similar pharmacokinetics in different patient populations. Since both are substrates, co-administration of rivaroxaban and apixaban with strong cytochrome P450 (CYP) 3A4 and permeability glycoprotein (P-gp) inhibitors and inducers can result in substantial changes in plasma concentrations due to altered clearance rates; consequently, their concomitant use is contraindicated and caution is required when used concomitantly with strong CYP3A4 and P-gp inducers. Although parenteral oral direct thrombin inhibitors (DTIs), such as argatroban and bivalirudin, have been on the market for years, DTIs such as dabigatran are novel synthetic thrombin antagonists. Dabigatran etexilate is a low-molecular-weight non-active pro-drug that is administered orally and converted rapidly to its active form, dabigatran—a potent, competitive and reversible DTI. Dabigatran has an advantage over the indirect thrombin inhibitors, unfractionated heparin and low-molecular-weight heparin, in that it inhibits free and fibrin-bound thrombin. The reversible binding of dabigatran may provide safer and more predictable anticoagulant treatment than seen with irreversible, non-covalent thrombin inhibitors, e.g. hirudin. Dabigatran shows a very low potential for drug–drug interactions. However, co-administration of dabigatran etexilate with other anticoagulants and antiplatelet agents can increase the bleeding risk. Although the new agents are pharmacologically better than VKAs—particularly in terms of fixed dosing, rapid onset of action, no INR monitoring and lower risk of drug interactions—there are some differences between them: the bioavailability of dabigatran is lower than rivaroxaban and apixaban, and so the dabigatran dosage required is higher; lower protein binding of dabigatran reduces the variability related to albuminaemia. The risk of metabolic drug–drug interactions also appears to differ between OACs: VKAs > rivaroxaban > apixaban > dabigatran. The convenience of the new OACs has translated into improvements in efficacy and safety as shown in phase III randomized trials. The new anticoagulants so far offer the greatest promise and opportunity for the replacement of VKAs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
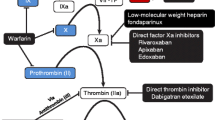
Oral anticoagulants (OACs) are indicated for the long-term prevention of thromboembolic disorders in several cardiovascular diseases, including atrial fibrillation (AF), acute myocardial infarction, venous thromboembolism (VTE) and in patients with mechanical heart valves. For several decades vitamin K antagonists (VKAs), e.g. warfarin, were the only OACs available in clinical practice. More recently, efforts have been made in research to develop pharmacological agents with more specific mechanisms of action than VKAs. Various anticoagulants act at different points in the anticoagulation cascade. Numerous new drugs directly inhibiting Factor Xa (FXa) or thrombin are approved, or are in late phase of approval, in some indications (Fig. 1).

Point of action of oral anticoagulants in the coagulation cascade. TF tissue factor, VKA vitamin K antagonist
This topic has been extensively reviewed elsewhere; therefore, the objective of this review is to compare the pharmacology of OACs currently in clinical use, focusing on the potential benefits of new drugs in comparison with VKAs, and to provide the clinician with tools for their appropriate use.
2 Vitamin K Antagonists
VKAs were discovered more than 80 years ago following the development of fatal bleeding in cattle that had eaten spoiled hay made from sweet clover. Dicoumarol was subsequently identified as a cause of these haemorrhagic complications by inhibiting the synthesis of vitamin K-dependent clotting factors [1–5].
Racemic warfarin is the principal VKA used worldwide, whereas phenprocoumon and acenocoumarol are VKAs widely used in many European countries. The VKAs in clinical use are structurally derived from 4-hydroxycoumarin and share a common mechanism of action in that they non-competitively inhibit the vitamin K epoxide reductase complex subunit 1 (VKORC1), which is essential in the recycling of vitamin K in the liver (Fig. 2) [6]. As vitamin K serves as a co-factor in the activation of clotting factors II, VII, IX and X, the inhibition of its recycling results in strong anticoagulation activity. On the other hand, vitamin K also serves as a co-factor for the anticoagulant proteins C, S and Z [7, 8], which also affects the regulation of the procoagulant–anticoagulant system.

The vitamin K cycle. Reproduced from Stehle et al. [6] with permission from Springer International Publishing AG (© Adis Data Information BV 2008. All rights reserved.). NAD(P) nicotine amide dinucleotide phosphate, NAD(P)H nicotine amide dinucleotide phosphate hydrogen, R aliphatic side chain, VKORC1 vitamin K epoxide reductase complex subunit 1
From a chemical point of view (Fig. 3), each drug has a single, chiral centre that gives rise to two different enantiomeric forms, of which the S-form is approximately two- to fivefold more potent than its R counterpart [9–12].

Structure of vitamin K antagonist anticoagulants
2.1 Pharmacokinetics
The comparative pharmacokinetics of VKAs have been extensively reported elsewhere [6, 13]. A summary of the key pharmacokinetic parameters of VKAs is shown in Table 1 [13].
After oral administration, VKAs are rapidly and extensively absorbed from the stomach and small intestine with essentially complete oral bioavailability, except for S-acenocoumarol, which undergoes extensive first-pass metabolism [14]. Although all VKAs are highly protein bound in human plasma (>98 %) [15], acenocoumarol has higher (almost double) apparent volumes of distribution than warfarin and phenprocoumon (Table 1) [13]. Since only the free fraction is pharmacologically active, variation in albuminaemia may affect the activity.
All three drugs are almost completely metabolised in specific pathways depending on the drug and enantiomeric form [6]. The elimination half-lives (t½) of phenprocoumon, acenocoumarol and warfarin range between 110–130, 1.8–6.6 and 24–58 h, respectively [13]. Reduced clearance by the cytochrome P450 (CYP) enzymes involved in phenprocoumon hydroxylation and enterohepatic recycling of conjugated phenprocoumon are probably the main cause of the longer t½ of phenprocoumon [13].
Hydroxylation and reduction processes in humans account for the metabolic clearance of warfarin of approximately 80 % in urine and 20 % in faeces. CYP2C9 is the main enzyme involved in warfarin metabolism and which mainly (>90 %) metabolises S-warfarin. The R-enantiomer is hydroxylated by CYP1A2, CYP2C8, CYP2C19 and CYP3A4. Furthermore, CYPs also facilitate the formation of dehydrowarfarin, but not the reduction of warfarin to alcohols [6]. In contrast, CYP2C9 hydroxylates all of the S-form of acenocoumarol and approximately 60 % of the R-enantiomer. The 6- and 7-hydroxylated metabolites and trace amounts of the parent compound are then excreted in their non-conjugated form via the kidneys [6].
CYP2C9 and CYP3A4 are the major enzymes responsible for the metabolism of phenprocoumon. In vitro, approximately 60 and 40 % of metabolites are formed by CYP2C9 and CYP3A4, respectively [6].
Numerous drug interactions with VKAs have been reported [13, 16]. These may occur by different mechanisms, including decreased absorption owing to an interruption of enterohepatic recycling, displacement from the protein-binding site and by inhibition or induction of metabolism. A complete listing of these data is beyond the scope of this review, but all inhibitors or inducers of CYP2C9 may affect the blood concentrations of VKAs.
2.2 Clinical Considerations
Of note, any conclusions on the efficacy and safety of warfarin may not be transferred directly to the other VKAs, because clinical studies with phenprocoumon and acenocoumarol are often lacking. However, these clinical considerations may be common to all VKAs.
VKAs are the orally active anticoagulants that are licensed for long-term use worldwide. The advantages of VKAs are their very low acquisition cost and the large experience in their management among physicians; however, they have important limitations that may neutralize any economic advantage.
VKAs have a slow onset of action and a narrow therapeutic window. They also have an unpredictable anticoagulant effect that results from multiple food and drug interactions, as well as genetic polymorphisms, that affect both the drug metabolism (CYP2C9) and the target of action (VKORC1). It is well known that CYP2C9 and VKORC1 polymorphisms have a strong impact on the responsiveness of warfarin and, to a lesser extent, of acenocoumarol and phenprocoumon, allowing derivation of their average effects on dose requirements [6]. However, the use of this information, which may reduce the adverse effects of warfarin in the treatment of patients, is still a rare exception.
In addition, non-genetic factors such as age, bodyweight, body surface area, sex, drug interactions, disease conditions and variable dietary intake of vitamin K can also contribute to drug use variation [17–21]. Patients must take dietary precautions, and prescribers must take special care when modifying concomitant drug therapy [22].
The unpredictable anticoagulant effects require routine coagulation monitoring and dose adjustment to maintain the international normalized ratio (INR) within the target range [23]. Furthermore, because VKAs have a slow onset of action, patients who require an immediate anticoagulant effect will require bridging therapy with a rapidly acting agent (e.g. low-molecular weight heparin) [23]. However, dietary precautions, dosing adjustments, monitoring and bridging therapy are inconvenient for patients, add to the cost of care and may be to blame for under-use or inappropriate use of VKA therapy [22]. Even when VKAs are appropriately used, the INR is frequently outside of the therapeutic range, leading to reduced anticoagulation or increased bleeding risk [24]. Poor anticoagulant control in patients with AF receiving VKA therapy for stroke prevention doubles the frequencies of stroke, major bleeding and death compared with those achieving good control [25].
Any conclusions on the efficacy of warfarin may not directly be transferred to the other VKAs, because clinical studies with phenprocoumon and acenocoumarol are often lacking.
A major requirement in management of thrombotic risk is an effective and safe replacement for VKAs—one that has a more rapid onset of action, a low potential for food and drug interactions, and a predictable anticoagulant effect that obviates the need for routine coagulation monitoring.
3 Factor Xa Antagonists
Two oral FXa inhibitors are reviewed here: rivaroxaban, which is approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA), and apixaban, which is approved by the EMA and is expected to be approved by the FDA at the beginning of 2013. Other FXa inhibitors include edoxaban (which was approved for use in Japan in April 2011), and otamixaban, betrixaban, DX-9065a, SR123781A, LY517717 and GW-813893, which are in earlier stages of development [26].
3.1 Rivaroxaban
Rivaroxaban is a potent, oral direct inhibitor of FXa. Rivaroxaban prevents thrombin generation inhibiting FXa by binding directly to the active sites of the serine endopeptidase. It has been shown to inhibit free FXa, FXa already bound in the prothrombinase complex, as well as clot-bound FXa. At the molecular level, the action of rivaroxaban is governed by chlorothiophene and morpholinone moieties attached to a central oxazolidinone ring, which bind with high affinity to the S1 and S4 pockets of FXa (Fig. 4) [27, 28].

Chemical structure of new oral anticoagulants. The morpholinone residue of rivaroxaban (a) binds the S4 pocket and the chlorothiophene moiety binds the S1 pocket of Factor Xa. [28] p-Methoxyphenyl of apixaban (b) binds the S1 pocket and the aryl-lactam moiety binds the S4 pocket of Factor Xa. [51] Dabigatran (c) specifically binds thrombin on the S and P pockets. The D pocket is occupied by a pyridine ring [102]
In preclinical studies, rivaroxaban dose-dependently inhibited free FXa in human plasma. The inhibition constant (Ki) for FXa in human plasma was 0.4 nmol/L; the rivaroxaban concentration required to inhibit 50 % of enzyme activity (IC50) was 21 nmol/L [29]. Rivaroxaban is specific for FXa and does not inhibit related serine proteases including thrombin, trypsin, plasmin, Factor VIIa, Factor IXa, Factor XIa, urokinase and activated protein C (IC50 >20,000 nmol/L) [29]. Plasma clotting times in human plasma were increased in a dose-dependent manner with rivaroxaban, with a concentration required to double prothrombin time (PT) and activated partial thromboplastin time (aPTT) of 0.23 and 0.69 mmol/L, respectively [29]. No direct effects of rivaroxaban on platelets have been demonstrated [26]. However, while rivaroxaban does not affect platelet aggregation in platelet-rich plasma, it potently inhibits tissue factor-induced platelet aggregation indirectly, by inhibiting thrombin generation [30].
Although the clinical relevance of these findings remains to be established, the effects of co-administration of rivaroxaban with naproxen [31], aspirin [32] and clopidogrel [33] have been examined in healthy volunteers. Naproxen and aspirin had no clinically relevant effect on bleeding times compared with rivaroxaban alone, while the combination of clopidogrel and rivaroxaban significantly prolonged the mean bleeding time compared with rivaroxaban alone (least-squares mean increase from baseline of 3.77-fold with combination therapy versus 1.13-fold with rivaroxaban alone [31–33]).
3.1.1 Pharmacokinetics
The pharmacokinetics of rivaroxaban have been evaluated in healthy young male subjects (aged 19–45 years) and elderly subjects (60–76 years) at single oral doses of up to 80 mg and multiple doses of up to 30 mg twice daily [34, 35]. Rivaroxaban reached maximum (peak) plasma concentrations (Cmax) 2–4 h after oral administration; the relative bioavailability of a tablet formulation versus a solution formulation in healthy volunteers is reported to be about 80 %. Dose-proportional increases in the area under the concentration–time curve (AUC) are observed, and there is no significant drug accumulation when steady-state conditions are achieved [35].
When co-administered with food, the time to Cmax (tmax) for rivaroxaban increased from 2.75 to 4 h, with an increase in the Cmax and AUC of 30–40 % [36]. Co-administration with food is associated with reduced inter-patient variability, thereby increasing the predictability of rivaroxaban plasma concentrations [36]. The Cmax is unaffected when rivaroxaban is given to subjects weighing about 120 kg, but a 24 % increase in the Cmax is seen when the drug is given to those weighing ≤50 kg [37].
When administering the same total daily dose, Cmax values are higher and minimum concentration values are lower with once-daily dosing than with twice-daily dosing in patients undergoing total hip replacement or receiving treatment for acute proximal deep vein thrombosis (DVT) [38, 39]. However, since the 90 % confidence intervals overlap, a concentration–effect relationship should be evaluated. Plasma protein binding is 92–95 %, with serum albumin the predominant binding component and the apparent volume of distribution of rivaroxaban at steady state is about 50 L [40].
Rivaroxaban has a terminal elimination half-life (t½β) of 5–9 h in healthy young subjects [35, 41], and 11–13 h in elderly subjects due to normal age-related renal function decline [34, 42]. In individuals with mild (creatinine clearance [CLCR] 50–80 mL/min), moderate (CLCR 30–49 mL/min) or severe (CLCR 15–29 mL/min) renal impairment, rivaroxaban plasma concentrations (AUC) were increased 1.4-, 1.5- and 1.6-fold, respectively, compared with healthy controls [42].
Rivaroxaban is metabolized via CYP enzymes (CYP3A4 and CYP2C8), as well as CYP-independent mechanisms, with the major sites of biotransformation being the oxidative degradation of the morpholinone moiety and hydrolysis of the amide bonds [43]. Approximately 66 % of ingested rivaroxaban is excreted via the kidneys and the remainder excreted in the faeces as unchanged drug. Intestinal excretion of rivaroxaban appears to be mediated, at least in part, by permeability glycoprotein (P-gp), because potent P-gp inhibitors increase rivaroxaban plasma concentration [44]. Of rivaroxaban excreted in the urine, 30–40 % is unchanged drug excreted via a combination of glomerular filtration and tubular secretion, and 60–70 % is metabolised [44].
The pharmacokinetic profile of rivaroxaban in healthy subjects is not substantially affected by age or sex [45]. Likewise, its profile is unaffected by mild impairment in hepatic function (Child-Pugh class A) [46] or extremes in bodyweight [37]. In subjects with moderate hepatic impairment (Child-Pugh class B), the AUC is twofold higher than in controls, with an associated moderate increase in inhibition of FXa activity and prolongation of the PT in these subjects [46].
As rivaroxaban is a substrate of CYP and P-gp, co-administration of rivaroxaban with strong CYP3A4 and P-gp inhibitors ketoconazole or ritonavir led to a 2.6- and 2.5-fold increase in mean rivaroxaban AUC and a 1.7- and 1.6-fold increase in mean rivaroxaban Cmax, respectively, with significant increases in pharmacodynamic effects that could result in increased bleeding risk [47]. The use of rivaroxaban is therefore contraindicated in patients receiving concomitant treatment with strong inhibitors of both CYP3A4 and P-gp [26].
Active substances strongly inhibiting only one of the rivaroxaban elimination pathways, either CYP3A4 or P-gp, are expected to increase rivaroxaban concentrations to a lesser extent. Co-administration of the strong CYP3A4 inhibitor and moderate P-gp inhibitors clarithromycin and erythromycin, for example, led to small and clinically insignificant increases in mean rivaroxaban AUC and Cmax [47].
Clopidogrel and rivaroxaban did not show a pharmacokinetic interaction when co-administered, but increased bleeding time not correlated to platelet aggregation was observed in some patients [33]. Care needs to be taken if patients are treated concomitantly with NSAIDs (including naproxen and aspirin) and platelet aggregation inhibitors because these medicinal products typically increase the bleeding risk [31–33].
Co-administration of rivaroxaban with the strong CYP3A4 inducer rifampicin led to an approximate 50 % decrease in mean rivaroxaban AUC, with parallel decreases in its pharmacodynamic effects [40]. The concomitant use of rivaroxaban with other strong CYP3A4 inducers (e.g. phenytoin, carbamazepine, phenobarbital or St John’s wort) may also lead to reduced rivaroxaban plasma concentrations and so they should be co-administered with caution [40].
3.1.2 Clinical Considerations
Rivaroxaban is generally administered once daily, although the drug is given twice daily for the initial treatment of patients with VTE [40].
The pharmacodynamic profile of rivaroxaban in healthy subjects reveals a linear correlation between the PT and the plasma concentration of rivaroxaban [35, 48, 49]. The maximum prolongations of the PT, aPTT and HepTest (a clot-based assessment measuring heparin in plasma) are dose dependent and follow the same time profiles as the pharmacokinetic time curves, with some prolongation observed up to 24 h after dosing [35, 41]. In two large, phase IIb, dose-finding trials investigating rivaroxaban for the treatment of proximal DVT or of patients undergoing hip replacement surgery [38, 50], clinically relevant plasma concentrations of rivaroxaban correlated linearly with the PT, suggesting that the PT may provide a useful assessment of drug exposure. Due to the linear correlation between plasma concentration and PT, care should be taken in elderly patients or in patients that are treated concomitantly with drugs affecting the exposure of rivaroxaban in order to avoid increased bleeding risk. Careful monitoring of PT should be performed in these ‘at risk’ patients [40].
In renal patients, the pharmacokinetic changes translated into more pronounced corresponding increases in pharmacodynamic effects: in individuals with mild, moderate and severe renal impairment, the overall inhibition of FXa activity was increased by a factor of 1.5, 1.9 and 2.0, respectively, compared with healthy volunteers; prolongation of PT was similarly increased by a factor of 1.3, 2.2 and 2.4, respectively [42]. There are no data in patients with CLCR <15 mL/min. Due to the high plasma protein binding of rivaroxaban, it is not expected to be dialysable and so its use is not recommended in patients with CLCR <15 mL/min, and the drug should be used with caution in patients with CLCR 15–29 mL/min [40].
3.2 Apixaban
Apixaban is a pyrazole derivative small-molecule, selective FXa inhibitor. The molecular formula for apixaban is C25H25N5O4; molecular weight 459.5 kDa [51]. Similarly to rivaroxaban, apixaban binds FXa at two sites (Fig. 4). Apixaban is a highly selective inhibitor of human FXa (apparent dissociation constant [Kd] 0.08 nmol/L); activated protein C, Factors IXa and VIIa, and thrombin were not affected [52]. Platelet aggregation was not altered by apixaban [53] and apixaban 10 mmol/L had no effect on platelet aggregation in response to adenosine diphosphate, gamma-thrombin or collagen stimulation [52]. Like rivaroxaban, apixaban is an inhibitor of FXa regardless of whether it is prothrombinase bound, associated with a clot or free FXa [54]. In platelet-poor human plasma, apixaban prolongs clotting times in a concentration-dependent manner: the PT, aPTT and HepTest were doubled by apixaban 3.6, 7.4 and 0.4 mol/L, respectively [55].
Apixaban inhibits thrombin generation in vitro [56], doubling the lag time and the peak thrombin concentration at 200 and 300 nmol/L, respectively. It reduces the maximum thrombin generation rate and the peak thrombin concentration, with IC50 values of 35 and 70 nmol/L, respectively.
Apixaban was as effective as fondaparinux and warfarin in preventing thrombosis at doses that preserve haemostasis in rabbit models [55]. Furthermore, the addition of apixaban to aspirin or clopidogrel significantly improved antithrombotic activity with a moderate, but significant, increase in bleeding time [57]. Apixaban significantly enhanced the antithrombotic effects of heparin and enoxaparin when co-administered, with a significant increase in bleeding time (p < 0.05) [58].
3.2.1 Pharmacokinetics
Apixaban is rapidly absorbed, reaching Cmax approximately 3 h post-dose in healthy volunteers [59]. The oral bioavailability of apixaban is approximately 50 % in human volunteers. Steady-state concentrations are reached within 3 days, with a mean t½β of 12.7 ± 8.55 h in healthy adults [26, 59–61]. Apixaban has a low volume of distribution of approximately 21 L and is about 87 % protein bound [26]. The small volume of distribution is probably due to limited extravascular tissue distribution and not the result of extensive plasma protein binding [54].
The effects of bodyweight, sex, age and ethnicity on apixaban pharmacokinetics have been reported [60, 62, 63]. After a single oral dose of apixaban 10 mg administered to healthy males and females stratified by bodyweight, the Cmax and AUC were 30 and 20 % higher, respectively, in subjects weighing <50 kg compared with those weighing 65–85 kg, and 30 and 20 % lower, respectively, than subjects weighing >120 kg [63]. In another study, apixaban 20 mg administered as a single oral dose to healthy younger (18–40 years) and elderly (>65 years) males and females produced a 32 and 7 % higher Cmax and AUC, respectively, in the elderly than in the younger participants and 18 and 15 % higher Cmax and AUC, respectively, in women than in men [60]. The European summary of product characteristics for apixaban states that the pharmacokinetic profile is similar in Asians, African Americans and Caucasians [64].
Food has little or no effect on Cmax or AUC [64].
After oral administration, the drug is eliminated by multiple routes, with approximately 50 % recovered unchanged in the faeces and approximately 25 % excreted in the urine [59]. Apixaban is metabolized via O-demethylation and hydroxylation mainly by CYP3A4/5 with minor contributions from CYP1A2 and 2J2 [65]. Unchanged apixaban is the major component in human plasma with no active circulating metabolites present [59]. In incubations of human liver microsomes, apixaban did not show inhibition (IC50 values >20 µmol/L) with the probe substrates of CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 or 3A4/5. Moreover, apixaban did not show any induction effect at concentrations up to 20 µmol/L on CYP1A2, 2B6 and 3A4/5 in incubations with primary human hepatocytes [65].
Although apixaban is a substrate of transport protein P-gp, it does not appear to be an inducer or inhibitor, so does not affect the pharmacokinetics of co-administered P-gp substrates [66]. However, strong inhibitors and inducers of both P-gp and CYP3A4 may affect apixaban pharmacokinetics. Co-administration of apixaban with ketoconazole, for example, a strong inhibitor of both CYP3A4 and P-gp, led to substantial increases in mean apixaban AUC and Cmax [66]. Therefore, the use of apixaban is not recommended in patients receiving concomitant systemic treatment with strong CYP3A4 and P-gp inhibitors, such as ketoconazole, ritonavir, etc. [64]. Moderate inhibitors of CYP3A4 and/or P-gp are expected to have a smaller effect on apixaban pharmacokinetics. Diltiazem, a moderate CYP3A4 and a weak P-gp inhibitor, led to small increases in mean apixaban AUC and Cmax [66]. Naproxen (500 mg, single dose) an inhibitor of P-gp but not an inhibitor of CYP3A4, led to a 1.5- and 1.6-fold increase in mean apixaban AUC and Cmax, respectively [61, 64]. Therefore, no dose adjustment for apixaban is advised when co-administered with less potent inhibitors of CYP3A4 and/or P-gp [64].
Co-administration of apixaban with rifampicin, a strong CYP3A4 and P-gp inducer, results in a significant decrease in mean apixaban AUC and Cmax [67]. The concomitant use of apixaban with other strong CYP3A4 and P-gp inducers (e.g. phenytoin, carbamazepine, phenobarbital or St John’s wort) may also lead to reduced apixaban plasma concentrations, so, although no dose adjustment is required during concomitant therapy with such agents, strong CYP3A4 and P-gp inducers should be co-administered with caution [64, 67].
3.2.2 Clinical Considerations
Apixaban is dosed twice daily [64]. Like rivaroxaban, apixaban-induced increases in the aPTT, INR and modified PT closely tracked the plasma concentration–time profile. Care should be taken in patients that are treated concomitantly with drugs affecting apixaban exposure, in order to avoid increased bleeding risk. Monitoring of plasma concentrations of apixaban may be warranted in this circumstance [64].
4 Oral Direct Thrombin Inhibitors
Direct thrombin inhibitors (DTIs) are a new therapeutic class of synthetic, small molecules that interact directly with thrombin and block its interaction with its substrates leading to an antithrombotic effect. Parenteral DTIs, such as argatroban and bivalirudin, have been on the market for many years. Ximelagatran, the pro-drug of melagatran, was the first orally available DTI to undergo clinical evaluation. However, although ximelagatran was proven to be an effective OAC, prolonged therapy with ximelagatran resulted in idiosyncratic hepatic toxicity and it was subsequently withdrawn from the market in 2006 [68]. Dabigatran is the second oral DTI approved for clinical use and has proven to be an effective OAC without the hepatic toxicity seen with ximelagatran [69].
4.1 Dabigatran Etexilate
Dabigatran etexilate is a low-molecular weight non-active pro-drug that is administered orally and converted in the blood to its active form, dabigatran, a potent, competitive and reversible DTI [70]. Dabigatran binds to the active site of thrombin univalently, thereby inactivating both fibrin-bound and unbound (i.e. free) thrombin (Fig. 4) [69, 71, 72]. Indirect thrombin inhibitors, such as unfractionated heparin and low-molecular weight heparin, cannot inhibit fibrin-bound thrombin. Dabigatran, therefore, has an advantage over the heparins because it prevents bound thrombin from continuing to trigger thrombus expansion [73, 74]. By inhibiting thrombin, dabigatran prevents a cascade of events: conversion of fibrinogen into fibrin, positive feedback amplification of coagulation activation, cross-linking of fibrin monomers, platelet activation and inhibition of fibrinolysis [70].
Dabigatran inhibits human thrombin in a concentration-dependent manner, with a Ki of 4.5 nmol/L, and has been shown to be a competitive inhibitor of thrombin [75]. Dabigatran displays highly selective and rapid, but reversible, binding to thrombin. Reversible binding may contribute towards safer and more predictable anticoagulant treatment than has been observed with irreversible, non-covalent binding, as shown for the first DTI, hirudin [76]. Dabigatran, by inhibiting thrombin, potently inhibits platelet aggregation, with an IC50 similar to the Ki of thrombin [75]. Moreover, dabigatran inhibits tissue factor-induced thrombin generation in human platelet-poor plasma in a concentration-dependent manner, and decreased endogenous thrombin generation [75]. A similar pattern has been observed in prolongation of blood coagulation times by dabigatran in animal species and human plasma, with the thrombin time and ecarin clotting time being the most sensitive clotting assays, followed by the aPTT and the PT [75, 77].
The antithrombotic and anticoagulant effects of the pro-drug dabigatran etexilate were investigated in a rabbit model [78]. In the rabbit jugular vein, dabigatran etexilate reduced thrombus formation in a dose-dependent manner with a dose that produced a 50 % effective response of 4.65 mg/kg, demonstrating effective antithrombotic activity. Thrombus formation was almost maximally reduced with dabigatran etexilate within 1 h of pretreatment, indicating a fast onset of action and rapid conversion to active dabigatran [78]. The preclinical data showed that dabigatran was an effective antithrombotic agent.
4.1.1 Pharmacokinetics
After oral administration, dabigatran etexilate is rapidly absorbed and quickly and completely hydrolysed to its active moiety, dabigatran, by non-specific ubiquitous esterases in the gut mucosa, liver and plasma [79, 80]. The absolute bioavailability after oral administration of dabigatran etexilate is about 7 % [79]. This low absolute bioavailability is not problematic from a clinical point of view, because the recommended doses ensure adequate plasma concentrations with no unexpected accumulation of dabigatran after multiple dosing [69]. After oral administration of dabigatran etexilate, Cmax is reached within 0.5–2 h (average, 1.5 h) [81] and steady-state concentrations are achieved within 3 days after multiple-dose administration in healthy volunteers [69]. Steady-state plasma concentrations in patients with AF taking 150 mg twice daily were 180 ng/mL at peak and 90 ng/mL at trough [82].
Dabigatran etexilate is not metabolised by the CYP enzymes or other oxidoreductases. About 20 % is conjugated with glucuronic acid and excreted via the biliary system [83]. In patients with mild hepatic impairment, the AUC after a single oral dose of dabigatran etexilate was comparable with that in healthy control subjects, and the bioconversion of the pro-drug was only slightly reduced [84].
Up to 80 % of circulating unchanged dabigatran and small amounts of dabigatran glucuronides are excreted via the kidneys, the dominant elimination pathway [81]. Consequently, reduced kidney function results in elevated dabigatran plasma concentrations and a prolonged drug t½ [85]. The mean t½β of dabigatran after oral administration is about 8–10 h after a single dose, and ranges from 14 to 17 h after multiple doses [79]. In older healthy volunteers the t½β is about 12–14 h [81]. The t½β is increased to 28 h in patients with a CLCR of <30 mL/min [85].
Dabigatran shows a very low potential for drug–drug interactions and the absorption is not affected by food. However, as with other OACs, co-administration of dabigatran etexilate with other anticoagulants and antiplatelet agents should be approached with caution because of an increased risk of bleeding.
Dabigatran etexilate absorption is dependent on an acid environment; therefore, it is formulated together with tartaric acid to reduce its variability [69]. Because of this, dabigatran etexilate absorption is independent of gastrointestinal tract acidity [70] and, although co-administration of the proton pump inhibitor (PPI) pantoprazole was shown to reduce the bioavailability of dabigatran by approximately 28 % and increase inter-patient pharmacokinetic variability, particularly in females, dabigatran dose adjustment is not required when co-administered with a PPI [81, 86].
Co-administration of atorvastatin (CYP3A4), diclofenac (CYP2C9) or digoxin (P-gp) with dabigatran has been shown to have limited impact on dabigatran efficacy, is safe and well tolerated, and does not affect routine coagulation assays [69, 87].
Dabigatran etexilate, but not dabigatran, is a substrate for P-gp. Co-administering dabigatran etexilate with strong P-gp inhibitors (e.g. ketoconazole and verapamil) and inducers (e.g. rifampicin) can alter plasma concentrations of dabigatran by decreasing or increasing clearance via the gastrointestinal tract [69]. Therefore, co-administration of strong P-gp inducers or inhibitors is contraindicated in patients receiving dabigatran etexilate after orthopaedic surgery [70].
4.1.2 Clinical Considerations
Prolongation of the clotting time with dabigatran correlates with the plasma concentration–time curve, with a rapid onset of action without time delay in healthy younger and older adults and the reproducible pharmacokinetic and pharmacodynamic profiles translate into convenient twice-daily dosing, with predictable rapid efficacy and good safety profile, without the need for coagulation monitoring or dose adjustment [79, 81, 88].
Since dabigatran is predominantly renally excreted, dose reduction may need to be considered in patients with moderate renal impairment (including age-related decline in renal function) and dabigatran is contraindicated in patients with severe renal function (CLCR <30 mL/min) [85, 89].
Moderate hepatic impairment has been shown to have no effect on the anticoagulant activity or safety profile of dabigatran and no dose adjustments are required [84].
5 Comparing Oral Anticoagulants
New OACs for the prevention and management of venous and arterial thromboembolism have important advantages over VKAs. Their rapid onset of action and predictable anticoagulant effects, which obviate the need for routine coagulation monitoring, are the most important. In addition, the low inter-patient variability and low propensity for drug interactions are important considerations for long-term anticoagulant therapy.
The pharmacological superiority of new OACs compared with VKAs is shown in Table 2. Although the new agents are pharmacologically better than VKAs, allowing fixed dosing, rapid onset of action and no monitoring of INR, interesting differences can be found between them. The bioavailability of dabigatran is lower than rivaroxaban and apixaban; therefore, the dosage of dabigatran is higher. The lower protein binding of dabigatran could be beneficial because it reduces the risk of variability related to albuminaemia and interactions for displacement. The risk of metabolic drug–drug interactions also appears to differ between OACs, and is dramatically higher for VKAs than for newer drugs. However, newer drugs also show a propensity for drug interactions. Overall, the extent of drug–drug interactions generally occur in the following order: VKAs > rivaroxaban > apixaban > dabigatran [26].
Furthermore, the convenience of the new OACs compared with existing anticoagulants has translated into improvements in efficacy and safety as shown in phase III randomized trials of the new agents compared with warfarin in AF: RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) (dabigatran), ROCKET-AF (Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) (rivaroxaban) and ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation)-AF (apixaban) (Table 3) [90–92]. Dabigatran, which had superior efficacy to warfarin with a lower bleeding risk in the RE-LY trial, so far offers the greatest promise and opportunity for the replacement of VKAs [90]. The results of the three pivotal studies comparing new OACs with warfarin in non-valvular AF, when compared indirectly, show that all new anticoagulants are able to reduce the emergence of stroke or systemic embolism versus warfarin. However, only dabigatran 150 mg twice daily and apixaban 5 mg twice daily are significantly superior to warfarin, with hazard ratios of 0.65 and 0.79, respectively (Table 4). Moreover, dabigatran showed a better overall safety profile compared with warfarin [90]. Finally, an increase in the risk of myocardial infarction observed with dabigatran 150 mg twice daily compared with warfarin, which was seen in the original analysis of RE-LY [90], was not confirmed in revised analysis [93].
Phase III results for rivaroxaban are also promising and provide additional information on the benefits of this agent in this clinical setting [91].
The withdrawal of oral ximelagatran, an oral DTI, in 2006 due to hepatic toxicity [68] has heightened awareness about potential adverse effects of these new agents, particularly hepatotoxicity, and associated drug interactions [94]. None of the new OACs in advanced stages of development or in clinical practice have demonstrated evidence of liver toxicity [94].
Finally, we must point out that the main adverse effect of all OACs is bleeding, and the absolute frequency differences between OACs are low. However, the new anticoagulants, in all studies, show a frequency of bleeding always lower than warfarin (Table 3).
Other potential limitations of these novel drugs include the lack of specific antidotes in such circumstances that immediate reversal is required (severe bleeding, urgent surgery, etc.). Prothrombin complex concentrate (PCC) from human plasma, containing a high concentration of the procoagulation factors II, VII, IX and X, as well as the natural anticoagulants protein C and S and antithrombin, has been proposed as antidote [95]. Although PCC may have important clinical implications, its effect has yet to be confirmed in patients with bleeding events who are treated with the new anticoagulants. However, the t½ values of the new agents are relatively short versus warfarin, so the absence of antidotes may not be a real limitation in the clinical setting.
Routine monitoring of drug plasma concentrations is not required with the new anticoagulants; however, a simple assay for quantifying drug activity or plasma concentrations would be useful in patients with a thrombotic or bleeding event and could be used to assess compliance [96, 97]. Such testing would be useful for drugs such as rivaroxaban or apixaban when they are administered in combination with drugs with a potential to cause metabolic interactions.
6 Conclusions
New OACs that directly inhibit FXa or thrombin provide numerous benefits over older VKAs. Although VKAs are economical and very well characterized, they have important limitations such as slow onset of action, narrow therapeutic window and unpredictable anticoagulant effect that make their use problematic and expensive. As a result, many patients do not receive optimum anticoagulant therapy leading either to increased bleeding risk or to reduced anticoagulation effect and increased risk of thrombotic events. The new OACs, direct FXa and thrombin inhibitors, have a rapid onset of action, low potential for food and drug interactions, and predictable anticoagulant effect, thereby removing the need for routine monitoring. These characteristics of new OACs increase their overall efficacy and reduce the risk of severe adverse effects such as major bleeding. Among the new OACs, dabigatran, which inhibits factor II (thrombin), shows a different mechanism of action, leading to a potent, competitive and reversible anticoagulant effect. The reversible binding of dabigatran may provide safer and more predictable anticoagulant treatment than seen with irreversible OACs. Furthermore, its low protein binding results in minimal albuminaemia-related inter-patient variability. Of all the newer OACs, dabigatran also appears to have the lowest risk of drug–drug interactions.
Although a pharmacoeconomic evaluation is not within the scope of this review, we must consider that the new anticoagulants have much higher prices than the VKAs. However, considering the overall cost of both VKAs and monitoring of INR, resource consumption of new OACs is comparably lower. Two pharmacoeconomic analyses that took into account dabigatran [98, 99] and apixaban [100, 101] have concluded that both drugs are cost effective relative to warfarin for secondary stroke prevention in patients with AF.
Even if a limited number of studies are published, it seems quite clear that the pharmacological benefits and convenience of the new OACs have translated into improvements in efficacy and safety as shown in phase III randomized trials, opening a new era in which anticoagulant therapy is less problematic, less dangerous, less costly and more effective.
References
Almquist HJ, Mecchi E, Klose AA. Estimation of the antihaemorrhagic vitamin. Biochem J. 1938;32(11):1897–903.
Dam H, Schonheyder F. The occurrence and chemical nature of vitamin K. Biochem J. 1936;30(5):897–901.
Dam H, Schonheyder F, Tage-Hansen E. Studies on the mode of action of vitamin K. Biochem J. 1936;30(6):1075–9.
Link KP. The discovery of dicumarol and its sequels. Circulation. 1959;19(1):97–107.
Mann FD, Mann JD, Bollman JL. The coagulation defect of vitamin K deficiency compared with that caused by dicumarol. J Lab Clin Med. 1950;36(2):234–7.
Stehle S, Kirchheiner J, Lazar A, et al. Pharmacogenetics of oral anticoagulants: a basis for dose individualization. Clin Pharmacokinet. 2008;47(9):565–94.
Rost S, Fregin A, Koch D, et al. Compound heterozygous mutations in the gamma-glutamyl carboxylase gene cause combined deficiency of all vitamin K-dependent blood coagulation factors. Br J Haematol. 2004;126(4):546–9.
Sadler JE. Medicine: K is for koagulation. Nature. 2004;427(6974):493–4.
Breckenridge A, Orme ML. The plasma half lives and the pharmacological effect of the enantiomers of warfarin in rats. Life Sci. 1972;11(7):337–45.
Jahnchen E, Meinertz T, Gilfrich HJ, et al. The enantiomers of phenprocoumon: pharmacodynamic and pharmacokinetic studies. Clin Pharmacol Ther. 1976;20(3):342–9.
Meinertz T, Kasper W, Kahl C, et al. Anticoagulant activity of the enantiomers of acenocoumarol. Br J Clin Pharmacol. 1978;5(2):187–8.
Schmidt W, Jahnchen E. Stereoselective drug distribution and anticoagulant potency of the enantiomers of phenprocoumon in rats. J Pharm Pharmacol. 1977;29(5):266–71.
Ufer M. Comparative pharmacokinetics of vitamin K antagonists: warfarin, phenprocoumon and acenocoumarol. Clin Pharmacokinet. 2005;44(12):1227–46.
Thijssen HH, Drittij MJ, Vervoort LM, et al. Altered pharmacokinetics of R- and S-acenocoumarol in a subject heterozygous for CYP2C9*3. Clin Pharmacol Ther. 2001;70(3):292–8.
de Vries JX, Volker U. Determination of the plasma protein binding of the coumarin anticoagulants phenprocoumon and its metabolites, warfarin and acenocoumarol, by ultrafiltration and high-performance liquid chromatography. J Chromatogr. 1990;529(2):479–85.
Harder S, Thurmann P. Clinically important drug interactions with anticoagulants: an update. Clin Pharmacokinet. 1996;30(6):416–44.
Greenblatt DJ, von Moltke LL. Interaction of warfarin with drugs, natural substances, and foods. J Clin Pharmacol. 2005;45(2):127–32.
Hillman MA, Wilke RA, Caldwell MD, et al. Relative impact of covariates in prescribing warfarin according to CYP2C9 genotype. Pharmacogenetics. 2004;14(8):539–47.
Khan T, Wynne H, Wood P, et al. Dietary vitamin K influences intra-individual variability in anticoagulant response to warfarin. Br J Haematol. 2004;124(3):348–54.
Miao L, Yang J, Huang C, et al. Contribution of age, body weight, and CYP2C9 and VKORC1 genotype to the anticoagulant response to warfarin: proposal for a new dosing regimen in Chinese patients. Eur J Clin Pharmacol. 2007;63(12):1135–41.
Momary KM, Shapiro NL, Viana MA, et al. Factors influencing warfarin dose requirements in African-Americans. Pharmacogenomics. 2007;8(11):1535–44.
Eriksson BI, Quinlan DJ, Eikelboom JW. Novel oral factor Xa and thrombin inhibitors in the management of thromboembolism. Ann Rev Med. 2011;62:41–57.
Keeling D, Baglin T, Tait C, et al. Guidelines on oral anticoagulation with warfarin: fourth edition. Br J Haematol. 2011;154(3):311–24.
Bungard TJ, Ghali WA, Teo KK, et al. Why do patients with atrial fibrillation not receive warfarin? Arch Intern Med. 2000;160(1):41–6.
White HD, Gruber M, Feyzi J, et al. Comparison of outcomes among patients randomized to warfarin therapy according to anticoagulant control: results from SPORTIF III and V. Arch Intern Med. 2007;167(3):239–45.
Eriksson BI, Quinlan DJ, Weitz JI. Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor xa inhibitors in development. Clin Pharmacokinet. 2009;48(1):1–22.
Laux V, Perzborn E, Kubitza D, et al. Preclinical and clinical characteristics of rivaroxaban: a novel, oral, direct factor Xa inhibitor. Semin Thromb Hemost. 2007;33(5):515–23.
Roehrig S, Straub A, Pohlmann J, et al. Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide (BAY 59-7939): an oral, direct factor Xa inhibitor. J Med Chem. 2005;48(19):5900–8.
Perzborn E, Strassburger J, Wilmen A, et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—an oral, direct Factor Xa inhibitor. J Thromb Haemost. 2005;3(3):514–21.
Perzborn E, Kubitza D, Misselwitz F. Rivaroxaban. A novel, oral, direct factor Xa inhibitor in clinical development for the prevention and treatment of thromboembolic disorders. Hamostaseologie. 2007;27(4):282–9.
Kubitza D, Becka M, Mueck W, et al. Rivaroxaban (BAY 59-7939)—an oral, direct Factor Xa inhibitor—has no clinically relevant interaction with naproxen. Br J Clin Pharmacol. 2007;63(4):469–76.
Kubitza D, Becka M, Mueck W, et al. Safety, tolerability, pharmacodynamics, and pharmacokinetics of rivaroxaban—an oral, direct factor Xa inhibitor—are not affected by aspirin. J Clin Pharmacol. 2006;46(9):981–90.
Kubitza D, Becka M, Mueck W, et al. Co-administration of rivaroxaban—a novel, oral, direct Factor Xa inhibitor—and clopidogrel in healthy subjects [abstract no. P1272]. Eur Heart J. 2007;28(Suppl 1):189.
Kubitza D, Becka M, Roth A, et al. Dose-escalation study of the pharmacokinetics and pharmacodynamics of rivaroxaban in healthy elderly subjects. Curr Med Res Opin. 2008;24(10):2757–65.
Kubitza D, Becka M, Wensing G, et al. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct Factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharm. 2005;61(12):873–80.
Kubitza D, Becka M, Zuehlsdorf M, et al. Effect of food, an antacid, and the H2 antagonist ranitidine on the absorption of BAY 59-7939 (rivaroxaban), an oral, direct factor Xa inhibitor, in healthy subjects. J Clin Pharmacol. 2006;46(5):549–58.
Kubitza D, Becka M, Zuehlsdorf M, et al. Body weight has limited influence on the safety, tolerability, pharmacokinetics, or pharmacodynamics of rivaroxaban (BAY 59-7939) in healthy subjects. J Clin Pharmacol. 2007;47(2):218–26.
Mueck W, Borris LC, Dahl OE, et al. Population pharmacokinetics and pharmacodynamics of once- and twice-daily rivaroxaban for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Haemost. 2008;100(3):453–61.
Mueck W, Lensing AW, Agnelli G, et al. Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep-vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50(10):675–86.
Bayer Pharma AG. Rivaroxaban summary of product characteristics. 2008. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000944/WC500057108.pdf. Accessed 1 Dec 2012.
Kubitza D, Becka M, Voith B, et al. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther. 2005;78(4):412–21.
Kubitza D, Becka M, Mueck W, et al. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct Factor Xa inhibitor. Br J Clin Pharmacol. 2010;70(5):703–12.
Lang D, Freudenberger C, Weinz C. In vitro metabolism of rivaroxaban, an oral, direct factor Xa inhibitor, in liver microsomes and hepatocytes of rats, dogs, and humans. Drug Metab Dispos. 2009;37(5):1046–55.
Gross PL, Weitz JI. New anticoagulants for treatment of venous thromboembolism. Arterioscler Thromb Vasc Biol. 2008;28(3):380–6.
Kubitza D, Becka M, Mueck W, et al. The effect of extreme age, and gender on the pharmacology and tolerability of rivaroxaban: an oral, direct factor Xa inhibitor. Blood. 2006;108(11):271–2.
Halabi A, Kubitza D, Zuehlsdorf M, et al. Effect of hepatic impairment on the pharmacokinetics, pharmacodynamics and tolerability of rivaroxaban: an oral, direct factor Xa inhibitor [abstract no. P-M-635]. J Thromb Haemost 2007 5(Suppl 2).
Gnoth MJ, Buetehorn U, Muenster U, et al. In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J Pharmacol Exp Ther. 2011;338(1):372–80.
Mueck W, Becka M, Kubitza D, et al. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor xa inhibitor—in healthy subjects. Int J Clin Pharmacol Ther. 2007;45(6):335–44.
Mueck W, Eriksson BI, Bauer KA, et al. Population pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor Xa inhibitor—in patients undergoing major orthopaedic surgery. Clin Pharmacokinet. 2008;47(3):203–16.
Agnelli G, Gallus A, Goldhaber SZ, et al. Treatment of proximal deep-vein thrombosis with the oral direct factor Xa inhibitor rivaroxaban (BAY 59-7939): the ODIXa-DVT (Oral Direct Factor Xa Inhibitor BAY 59-7939 in patients with acute symptomatic deep-vein thrombosis) study. Circulation. 2007;116(2):180–7.
Wong PC, Pinto DJ, Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis. 2011;31(4):478–92.
Pinto DJ, Orwat MJ, Koch S, et al. Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem. 2007;50(22):5339–56.
Wong PC, Jiang X. Apixaban, a direct factor Xa inhibitor, inhibits tissue-factor induced human platelet aggregation in vitro: comparison with direct inhibitors of factor VIIa, XIa and thrombin. Thromb Haemost. 2010;104(2):302–10.
He K, Luettgen JM, Zhang D, et al. Preclinical pharmacokinetics and pharmacodynamics of apixaban, a potent and selective factor Xa inhibitor. Eur J Drug Metab Pharm. 2011;36(3):129–39.
Wong PC, Crain EJ, Xin B, et al. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost. 2008;6(5):820–9.
Barrett YC, Wang Z, Frost C, et al. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost. 2010;104(6):1263–71.
Wong PC, Watson CA, Crain EJ. Arterial antithrombotic and bleeding time effects of apixaban, a direct factor Xa inhibitor, in combination with antiplatelet therapy in rabbits. J Thromb Haemost. 2008;6(10):1736–41.
Wong P, Watson C, Knabb R, Crain E. The combination of apixaban, a direct factor Xa inhibitor, with heparin or enoxaparin in rabbits elicits additive antithrombotic effects, with low bleeding [abstract no. 933]. Annual Congress of the European Society of Cardiology (ESC) Munich; 30 Aug–3 Sep 2008.
Raghavan N, Frost CE, Yu Z, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37(1):74–81.
Frost CE, Nepal S, Barrett Y, et al. Effects of age and gender on the single-dose pharmacokinetics (PK) and pharmacodynamics (PD) of apixaban [abstract no. PP-MO-407]. J Thromb Haemost. 2009;7(Suppl 2):455.
Prom R, Spinler SA. The role of apixaban for venous and arterial thromboembolic disease. Ann Pharmacother. 2011;45(10):1262–83.
Song Y, Cui Y, Li T, et al. Apixaban pharmacokinetics and pharmacodynamics in healthy Chinese subjects [abstract no. 22]. J Clin Pharmacol. 2010;50:1062.
Upreti VV, Wang J, Barrett YC, et al. Effect of body weight on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor, in healthy subjects [abstract no. 16]. J Clin Pharmacol. 2010;50:1060.
BMS/Pfizer. Eliquis (apixaban) summary of product characteristics. 2011. http://www.eliquis.com/PDF/ELIQUIS%20%C2%AE%20(apixaban)%20SmPC.pdf. Accessed 1 Dec 2012.
Wang L, Zhang D, Raghavan N, et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–58.
Frost C, Wang J, Nepal S, et al. Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor [abstract no. 139]. J Clin Pharmacol. 2009;49:1123.
Vakkalagadda B, Frost C, Wang J, et al. Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa [abstract no. 143]. J Clin Pharmacol. 2009;49:1124.
European Medicines Agency Press Office. AstraZeneca withdraws its application for Ximelagatran 36-mg film-coated tablets. 2006. http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2010/02/WC500074073.pdf. Accessed 1 Dec 2012.
Eisert WG, Hauel N, Stangier J, et al. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol. 2010;30(10):1885–9.
Hankey GJ, Eikelboom JW. Dabigatran etexilate: a new oral thrombin inhibitor. Circulation. 2011;123(13):1436–50.
Huntington JA, Baglin TP. Targeting thrombin: rational drug design from natural mechanisms. Trends Pharmacol Sci. 2003;24(11):589–95.
van Ryn J, Hauel N, Waldman L, et al. Dabigatran inhibits both clot-bound and fluid-phase thrombin in vitro: comparison to heparin and hirudin [abstract no. 570]. Arterioscler Thromb Vasc Biol. 2008;28:e136–7.
Weitz JI, Hudoba M, Massel D, et al. Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J Clin Invest. 1990;86(2):385–91.
Maegdefessel L, Linde T, Krapiec F, et al. In vitro comparison of dabigatran, unfractionated heparin, and low-molecular-weight heparin in preventing thrombus formation on mechanical heart valves. Thromb Res. 2010;126(3):e196–200.
Wienen W, Stassen JM, Priepke H, et al. In-vitro profile and ex-vivo anticoagulant activity of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate. Thromb Haemost. 2007;98(1):155–62.
Markwardt F. Hirudin as alternative anticoagulant: a historical review. Semin Thromb Hemost. 2002;28(5):405–14.
Wienen W, Stassen JM, Priepke H, et al. Effects of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate, on thrombus formation and bleeding time in rats. Thromb Haemost. 2007;98(2):333–8.
Wienen W, Stassen JM, Priepke H, et al. Antithrombotic and anticoagulant effects of the direct thrombin inhibitor dabigatran, and its oral prodrug, dabigatran etexilate, in a rabbit model of venous thrombosis. J Thromb Haemost. 2007;5(6):1237–42.
Stangier J, Rathgen K, Stahle H, et al. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol. 2007;64(3):292–303.
Troconiz IF, Tillmann C, Liesenfeld KH, et al. Population pharmacokinetic analysis of the new oral thrombin inhibitor dabigatran etexilate (BIBR 1048) in patients undergoing primary elective total hip replacement surgery. J Clin Pharmacol. 2007;47(3):371–82.
Stangier J, Stahle H, Rathgen K, et al. Pharmacokinetics and pharmacodynamics of the direct oral thrombin inhibitor dabigatran in healthy elderly subjects. Clin Pharmacokinet. 2008;47(1):47–59.
van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2010;103(6):1116–27.
Ebner T, Wagner K, Wienen W. Dabigatran acylglucuronide, the major human metabolite of dabigatran: in vitro formation, stability, and pharmacological activity. Drug Metab Dispos. 2010;38(9):1567–75.
Stangier J, Stahle H, Rathgen K, et al. Pharmacokinetics and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor, are not affected by moderate hepatic impairment. J Clin Pharmacol. 2008;48(12):1411–9.
Stangier J, Rathgen K, Stahle H, et al. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet. 2010;49(4):259–68.
Stangier J, Eriksson BI, Dahl OE, et al. Pharmacokinetic profile of the oral direct thrombin inhibitor dabigatran etexilate in healthy volunteers and patients undergoing total hip replacement. J Clin Pharmacol. 2005;45(5):555–63.
Stangier J, Rathgen K, Stahle H, et al. Coadministration of dabigatran etexilate and atorvastatin: assessment of potential impact on pharmacokinetics and pharmacodynamics. Am J Cardiovasc Drugs. 2009;9(1):59–68.
Clemens A, Haertter S, Friedman J, et al. Twice daily dosing of dabigatran for stroke prevention in atrial fibrillation: a pharmacokinetic justification. Curr Med Res Opin. 2012;28(2):195–201.
Boehringer Ingelheim International GmbH. Pradaxa (dabigatran etexilate) summary of product characteristics. 2008. http://www.eliquis.com/PDF/ELIQUIS%20%C2%AE%20(apixaban)%20SmPC.pdf. Accessed 1 Dec 2012.
Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–51.
Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–91.
Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–92.
Connolly SJ, Ezekowitz MD, Yusuf S, et al. Newly identified events in the RE-LY trial. N Engl J Med. 2010;363(19):1875–6.
Eikelboom JW, Weitz JI. New anticoagulants. Circulation. 2010;121(13):1523–32.
Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation. 2011;124(14):1573–9.
Bounameaux H, Reber G. New oral antithrombotics: a need for laboratory monitoring. Against [comment]. J Thromb Haemost. 2010;8(4):627–30.
Weitz JI. New oral anticoagulants in development. Thromb Haemost. 2010;103(1):62–70.
McKeage K. Dabigatran etexilate: a pharmacoeconomic review of its use in the prevention of stroke and systemic embolism in patients with atrial fibrillation. Pharmacoeconomics. 2012;30(9):841–55.
Davidson T, Husberg M, Janzon M, Oldgren J, Levin LA. Cost-effectiveness of dabigatran compared with warfarin for patients with atrial fibrillation in Sweden. Eur Heart J. 2012 [epub ahead of print].
Kamel H, Easton JD, Johnston SC, Kim AS. Cost-effectiveness of apixaban vs warfarin for secondary stroke prevention in atrial fibrillation. Neurology. 2012;79(14):1428–34.
Lee S, Mullin R, Blazawski J, Coleman CI. Cost-effectiveness of apixaban compared with warfarin for stroke prevention in atrial fibrillation. PLoS One. 2012;7(10):e47473.
Hauel NH, Nar H, Priepke H, et al. Structure-based design of novel potent nonpeptide thrombin inhibitors. J Med Chem. 2002;45(9):1757–66.
Acknowledgments
No sources of funding were used to assist in the preparation of this review. The author declares that no conflicts of interest exist.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Scaglione, F. New Oral Anticoagulants: Comparative Pharmacology with Vitamin K Antagonists. Clin Pharmacokinet 52, 69–82 (2013). https://doi.org/10.1007/s40262-012-0030-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-012-0030-9