
Abstract
Background and Objective
Colchicine is commonly prescribed for gout. While minimally metabolized by the cytochrome P450 (CYP) 3A4 isoenzyme, colchicine is a substrate for P-glycoprotein (P-gp). Atorvastatin is metabolized primarily by CYP3A4 and is a P-gp inhibitor. Patients with gout often have dyslipidemia; therefore, the potential for co-administration of atorvastatin and colchicine exists. The objective of this study was to determine the effect of oral atorvastatin on the pharmacokinetics of a single, oral dose of colchicine.
Methods
Twenty-four healthy adult subjects were enrolled in this single-center, open-label, non-randomized, one-sequence, two-period drug–drug interaction study. On day 1, subjects received a single oral dose of colchicine 0.6 mg. After a 14-day washout, subjects received atorvastatin 40 mg once daily for 14 days followed by a single dose of colchicine 0.6 mg co-administered with atorvastatin 40 mg on day 28. Main outcome measures were colchicine maximum plasma concentration (C max), area under the plasma concentration–time curve (AUC) from time zero to the last measurable concentration (AUClast), and AUC from time zero to infinity (AUC∞), which were compared with and without concurrent atorvastatin.
Results
Colchicine AUClast, AUC∞, and C max increased by 27, 24, and 31 %, respectively, when co-administered with atorvastatin. Corresponding 90 % confidence intervals around the ratios were outside the established no-effect 80–125 % interval.
Conclusion
Increased colchicine exposure was observed after a single dose of colchicine was administered with steady-state atorvastatin. Additional studies with multiple dosing of both drugs are needed to further determine the clinical implications of these results.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Gout, a painful and often debilitating rheumatologic disorder, affects approximately 8.3 million adults in the United States [1]. Colchicine is one of the first-line options to prevent and treat gout flares [2]. Gout is often associated with a number of co-morbidities [3], such as dyslipidemia, which was recently reported in 27 % of patients with gout in a US managed care population [4]. Statins (HMG-CoA reductase inhibitors), such as atorvastatin, are commonly prescribed lipid-lowering drugs. Therefore, patients with gout and dyslipidemia may be co-prescribed colchicine and atorvastatin. Anticipating any drug–drug interaction is necessary for safe patient management as increased plasma colchicine concentrations may lead to severe and dangerous toxicity [5]. Cases of myopathy and rhabdomyolysis have been reported when colchicine was co-administered with a statin [6–12].
Increased exposure to colchicine can occur when co-administered with drugs that inhibit its metabolism or transport. Colchicine is minimally metabolized in the liver with less than 5 % metabolized by the cytochrome P450 (CYP) 3A4 isoenzyme [13, 14]. Colchicine is also a substrate for P-glycoprotein (P-gp) [15], a protein involved in the multidrug resistance transport system. Renal and hepatic excretion of colchicine involves P-gp-mediated efflux across membranes [16, 17]. Both enterohepatic recirculation and biliary excretion, mediated by P-gp, are postulated to be major routes of colchicine elimination [18, 19]. Toxicity can also be increased by drugs that inhibit the P-gp efflux pump and allow for more colchicine to be re-absorbed [5]. P-gp-mediated intestinal secretion and reabsorption/biliary recirculation of colchicine do occur, as evidenced by secondary peak plasma concentrations and excretion of unchanged colchicine in feces [13, 14]. Atorvastatin is also a direct P-gp inhibitor [20], and co-administration of atorvastatin with other drugs that are substrates/inhibitors for P-gp and CYP3A4, such as digoxin [21] and verapamil [22], significantly increased their bioavailability.
Administration of a single dose of colchicine with strong inhibitors of P-gp [cyclosporine (ciclosporin)] or CYP3A4 (clarithromycin, ketoconazole, ritonavir) increased systemic colchicine exposure by ~300 % [23]. As such, colchicine dose reductions are recommended for patients co-prescribed colchicine with strong P-gp or CYP3A4 inhibitors [5]. The goal of this drug–drug interaction study was to investigate the potential of atorvastatin to alter systemic concentrations of colchicine. The effect of multiple doses of oral atorvastatin (40 mg once daily for 14 days) on the pharmacokinetics and tolerability of a single oral dose of colchicine (0.6 mg) in healthy adult subjects under fasting conditions was tested.
2 Methods
2.1 Study Subjects
Healthy adults aged 18–45 years with a body mass index of 18–32 kg/m2 were eligible to enroll in this study (ClinicalTrials.gov registered study number NCT00960323). Health status was assessed based on medical history, physical examination, and routine laboratory tests including renal and hepatic function, vital signs, and electrocardiography. All subjects had to be non-smokers (including use of nicotine-containing products) for ≥6 months prior to the first dose. Women had to be surgically sterile (hysterectomy or bilateral oophorectomy) or had to have undergone bilateral tubal ligation ≥6 months before study entry.
Exclusion criteria included allergies to colchicine, atorvastatin, or any other statin; a history or presence of significant cardiovascular, pulmonary, hepatic, gallbladder, biliary tract, renal, hematologic, gastrointestinal, endocrine, immunologic, dermatologic, neurologic, or psychiatric disease; use of any drugs or substances known to inhibit CYP enzymes and/or P-gp in the 4 weeks before the first dose and throughout the study; a positive test result for HIV or hepatitis B or C at screening; history or evidence of alcoholism or drug abuse in the previous 2 years; active sexually transmitted disease; use of a special diet in the 4 weeks before the first dose; difficulty in fasting or consuming standardized meals; hemoglobin concentration <11.5 g/dL; inadequate venous access for repeated venipuncture; blood donation of 50–499 mL within 4 weeks and >499 mL within 8 weeks before the first dose; blood donation of >500 mL within 2 weeks, >1,500 mL in 6 months, or >2,500 mL in 1 year at completion of the study; donation of plasma in the 4 weeks before the first dose; participation in any other clinical trial within 4 weeks before the first dose; and pregnancy, lactation, or childbearing potential.
2.2 Study Design
All subjects participated in the study at a single US clinical study center (Cetero Research, Fargo, ND, USA; phase I facility), and the study protocol was approved by the ethics committee of the Institutional Review Board of the PRACS Institute, Ltd. All subjects provided written informed consent before participation in the study, which was conducted in accordance with the US Code of Federal Regulations and International Conference on Harmonization Guidelines for Good Clinical Practice and adhered to the ethical principles of the Declaration of Helsinki.
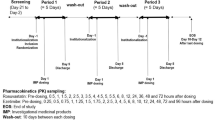
This was a phase I, open-label, non-randomized, one-sequence, drug-interaction study (Fig. 1) wherein a single 0.6 mg dose of colchicine (Colcrys® Tablets; Takeda Pharmaceuticals USA, Deerfield, IL, USA) was administered alone or co-administered with steady-state atorvastatin calcium (Lipitor® Tablets; Pfizer Inc., New York, NY, USA) 40 mg once daily for 14 days. Following a 4-week screening period (days −28 to −2), subjects were given colchicine 0.6 mg on day 1 (period 1). After completing a 14-day washout period, subjects started period 2 and received atorvastatin 40 mg once daily, in the morning after an overnight fast, for a total of 14 days (days 15–27). On day 28, subjects received a single dose of colchicine 0.6 mg in combination with atorvastatin 40 mg. Subjects were admitted to the clinical facility for ~36 h (~12 h before until ~24 h after dosing) on colchicine-dosing days, administered alone and in combination with atorvastatin [days −1 to 2 (period 1) and days 27–29 (period 2), respectively]. Doses of colchicine were administered after subjects completed a 10-h overnight fast; all doses of atorvastatin and/or colchicine were administered with 240 mL of water. Subjects received doses sequentially in pairs at 1-min intervals as a single cohort and in the same sequence during each study period. Compliance was confirmed by oral cavity and hand inspection. No food was permitted until 4.25 h after administration of colchicine, while clear fluids were permitted from 2 h post-dose.

Study design. AM morning
During the clinical confinement period from days −1 to 2 and 27–29, standardized meals, beverages, and a standardized snack were provided at 4.25, 10.25, and 14.25 h after administration of colchicine. Meals and snacks were identical during each study period and were free from grapefruit as well as xanthine- and caffeine-containing products. Colchicine and atorvastatin were administered in the morning of each day of dosing after a 10-h overnight fast. Water was allowed ad libitum starting 2 h after administration of colchicine. On days 15–27, subjects were not confined to the study unit and meals were not monitored.
Subjects were instructed not to take prescription or over-the-counter medications, herbal products, or suprapharmacologic doses of vitamins or supplements for 28 days before the first dose of study medication and throughout the duration of the study. They were also instructed to abstain from consuming products containing caffeine, xanthine, and alcohol for 48 h and to refrain from eating grapefruit or grapefruit-containing products for 14 days before the first study dose and throughout the study. They were requested to refrain from engaging in strenuous activities at any time during the confinement periods.
Subjects were free to withdraw from the study at any time for any reason. Furthermore, subjects could be withdrawn by the investigator in the event of an adverse event (AE) or laboratory abnormality, development of an illness that would preclude continuation in the study, or non-compliance.
2.3 Safety Assessments
Medical history, physical examination, vital signs, 12-lead electrocardiography, urinary drug screen, and routine laboratory tests were performed during the screening period (day −28 to −2). Medical history, vital signs, urinary drug screen, and routine laboratory tests were re-assessed at confinement to the clinic on the day before administration of colchicine (days −1 and 28) and at the time of discharge from the clinic at the end of the study (day 29). Blood pressure and heart rate were measured with the subjects in a seated position for ≥5 min before administration of colchicine and at 1 and 2 h post-dose.
Any undesirable sign, symptom, or medical condition occurring after the start of the study—whether reported spontaneously or in response to indirect questioning, or directly observed by physical examination, monitoring of vital signs, or clinical laboratory testing—was recorded by the investigator as an AE regardless of suspected relation to the study medication (coded using Medical Dictionary for Regulatory Activities version 10.1) and was graded by intensity (mild, moderate, or severe) and relationship to the study drug (unrelated, unlikely, possibly, or probably). Any serious AE was reported expeditiously.
2.4 Pharmacokinetic Measurements
Blood (6-mL aliquots) was taken by direct venipuncture at 0 (pre-dose) and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, and 24 h after administration of colchicine on days 1 and 28. Colchicine plasma concentrations were measured (Cetero Research, Fargo, ND, USA) using a validated liquid chromatography/tandem mass spectrometry (LC/MS/MS) method (API 5000TM LC/MS/MS System with a Turbo Ion SprayTM source; AB Sciex, Framingham, MA, USA). For this method, the between-batch accuracy and precision for colchicine was determined by analyzing three standard curves consisting of nine concentrations prepared in doublet in three separate batches, with four concentrations of quality controls (QCs). The accuracy of the colchicine standards and QCs were between 98.57 and 101.92 %, and 101.17 and 106.22 %, respectively, while precision was ≤5.60 and ≤6.38 %, respectively. Within-batch accuracy was assessed using six replicates of five concentrations of QC samples, and ranged from 96.75–109.17 %, while precision was ≤5.93 %. Analysis of plasma samples was partially blinded; information on the tubes did not include any reference to the treatment regimen.
To determine the plasma concentrations of colchicine, extracts were analyzed by LC/MS/MS. The plasma concentration of colchicine was ascertained by comparison with a standard curve (range 0.20–40 ng/mL). Standards and QC samples were incorporated into each batch analysis and the validity of each batch run was determined by assessing the accuracy of the standards and QC samples. All samples for each subject were analyzed together. The lower limit of quantification for colchicine was 0.2 ng/mL. Values below the lower limit of quantification were set to zero.
Pharmacokinetic parameters were determined using the WinNonlin® version 5.0.1 (Pharsight, Sunnyvale, CA, USA) using standard non-compartmental methods to determine the following pharmacokinetic parameters: maximum observed plasma concentration (C max); time to reach C max (t max); area under the plasma concentration-time curve (AUC) from time zero to the last measurable concentration (C last) calculated by the linear trapezoidal method (AUClast); apparent first-order terminal elimination rate constant (k el) calculated from a semi-log plot of the plasma concentration–time curve calculated by linear least-squares regression analysis using the maximum number of points in the terminal log-linear phase (e.g., ≥3 non-zero plasma concentration); AUC from time zero to infinity (AUC∞) calculated as AUClast + C last/k el; apparent first-order terminal elimination half-life (t ½) calculated as 0.693/k el; apparent total body clearance (CL/F) calculated as the dose/AUC∞; and apparent total volume of distribution (V d/F) calculated as the dose/(AUC∞ × k el).
Samples with significant deviation from protocol schedule times were not included in the analysis. Furthermore, data from subjects who experienced emesis within 3 h post-dose were excluded from analysis in accordance with current US FDA guidelines [24]; for immediate-release products, the duration of no emesis is twice the mean t max, which for colchicine is about 1.5 h [5].
2.5 Statistical Analysis
The sample size of 24 subjects used in this study was based on other published atorvastatin drug interaction studies, colchicine drug interaction studies, and other colchicine pharmacokinetic studies recently conducted by the sponsor [23]. A formal sample size calculation was not performed.
Descriptive statistics were used to summarize the pharmacokinetic parameter values. The ANOVA model, which included sequence, treatment (i.e., formulation), and period as fixed effects, and subject nested within sequence as a random effect, was performed on colchicine natural logarithms of C max, AUClast, and AUC∞ using an SAS® (SAS Institute, Cary, NC, USA) general linear model procedure. The effect of multiple doses of atorvastatin on the single-dose pharmacokinetic of colchicine was assessed via point estimates and 90 % confidence intervals (CIs) for the ratios (colchicine + steady-state atorvastatin/colchicine alone) of the central values of the C max and AUCs of colchicine. No effect of atorvastatin on the pharmacokinetic of colchicine was declared if the 90 % CIs for the ratios of the central values of the C max and AUCs of colchicine were within the no-effect range of 80–125 %, in accordance with FDA guidelines [24]. The Wilcoxon signed rank test statistic was used to analyze t max with a P value <0.05 considered as significant.
3 Results
Twenty-four subjects were enrolled and evaluated for safety. Of these, 23 had paired data that was used for pharmacokinetic analyses; one subject discontinued the study due to a scheduling conflict. All subjects received the first dose of study medication on 7 March 2009, and the last pharmacokinetic blood sample was collected on 4 April 2009. The demographic characteristics of the per-protocol population are summarized in Table 1. The majority of the study population was white (91.7 %) and male (91.7 %).
Plasma concentration-time curve profiles for colchicine alone versus colchicine and steady-state atorvastatin are shown in Fig. 2. Colchicine was rapidly absorbed when administered with and without atorvastatin. The plasma concentrations of colchicine 24 h post-dose were below the limit of quantification when administered as a single dose (in 1/24 patients) or in combination with atorvastatin (in 5/24 patients).

Mean colchicine plasma concentrations of single doses of colchicine 0.6 mg tablets (0–24 h) before and after multiple-dose atorvastatin (N = 23). Error bars represent the standard deviation
The mean pharmacokinetic parameters for colchicine alone and colchicine plus steady-state atorvastatin are summarized in Table 2. Exposure was higher when colchicine was co-administered with atorvastatin; mean C max values for colchicine alone and with steady-state atorvastatin were 2.02 and 2.49 ng/mL, respectively, while mean AUC∞ values were 9.59 and 11.04 ng·h/mL, respectively. Median t max was comparable at 1 h.
Point estimates [ratios of the central values for the test treatment (colchicine + atorvastatin) and the reference treatment (colchicine alone)] and 90 % CIs were calculated based on ANOVA models on natural logarithm–transformed C max, AUClast, and AUC∞ values. The rate (C max) and extent (AUC) of colchicine absorption increased by ~31 and ~24 %, respectively, when colchicine was administered with steady-state atorvastatin. The 90 % CIs for the ratios of the central values of C max, AUClast, and AUC∞ were outside the no-effect range of 80–125 %. The point estimates along with their 90 % CIs indicate that a drug interaction was likely present when colchicine and atorvastatin were co-administered (Table 3). The t max was not affected (P = 0.7899) by co-administration with atorvastatin.
AEs are summarized in Table 4. Seventeen treatment-emergent AEs (TEAEs) were reported by seven subjects (29.2 %) during the course of this study; seven of these were reported by four subjects (16.7 %) following the administration of colchicine alone, nine AEs were reported by six subjects (25.0 %) following the administration of atorvastatin alone, and one AE was reported (4.2 %) following the combination of colchicine and atorvastatin. Dizziness, fatigue, and cold sweats comprised nine of the 17 AEs; however, these symptoms were reported eight times by a single subject over the entire study and it was almost always related to phlebotomy. Excluding the events reported by this one subject reduces the overall reporting of AEs to nine events reported by six subjects; three subjects reported three AEs following administration of colchicine only, six AEs were reported by five subjects during administration of atorvastatin only, and no AEs were reported when colchicine and atorvastatin were administered concomitantly.
There were no discontinuations due to AEs, serious AEs, or deaths. All TEAEs were mild or moderate in intensity. There was a low incidence of AEs during the administration of a single dose of colchicine or during its co-administration with atorvastatin 40 mg at steady state [four and one subject(s), respectively] with no clear difference between the two administrations. As was expected, a greater number of subjects experienced a higher number and wider range of AEs during the 14-day administration of atorvastatin 40 mg. No consistent changes were reported during the study with respect to laboratory test values, vital signs, or physical findings. None of the treatment-emergent abnormalities were directly attributable to the study medications.
4 Discussion
A number of studies have reported AEs such as neuromyopathy and/or rhabdomyolysis during the co-administration of colchicine with various statins, including atorvastatin [6, 7], simvastatin [7–9, 25, 26], pravastatin [10, 27], and fluvastatin [11, 12]. Both colchicine and statins have the potential for myotoxicity; thus, it is often difficult to attribute the cause of the toxicity solely to colchicine, the statin, a combination of the two drugs, or any other co-administered drug in case reports when drug plasma concentrations are not determined. This study showed that when a single dose of colchicine was administered with steady-state atorvastatin, C max values of colchicine were increased by 31 % while systemic exposure increased by 24 % on average. The 90 % CIs for the ratios of the central values of C max, AUClast, and AUC∞ were 111–153 %, 109–149 % and 110–141 % respectively, which are outside the no-effect range of 80–125 %. The point estimates along with their 90 % CIs indicate that a drug interaction was likely when colchicine and atorvastatin were co-administered. The clinical impact of this increased exposure is unclear. Despite the increased exposure to colchicine, there were no apparent differences in AE profiles when colchicine was administered alone or with steady-state atorvastatin (Table 4). Additional studies with multiple doses of colchicine and atorvastatin are needed to determine if co-administration of these drugs in patients with gout, reflective of what would occur in a clinical setting, leads to increased rates of myopathic side effects.
Potential mediators for the interaction between colchicine and atorvastatin are CYP3A4 and/or P-gp. Several indications are against the involvement of CYP3A4 solely. While less than 5 % of colchicine is metabolized by the hepatic CYP3A4 [13, 14], atorvastatin is primarily metabolized by CYP3A4 [28] and may therefore compete with colchicine for the CYP3A4 enzyme. Atorvastatin, while orally administered as an acid, will shift to its lactone form in vivo, with both forms present roughly in equilibrium [28]. Both atorvastatin acid and lactone are metabolized primarily by CYP3A4 to two active metabolites. Atorvastatin is considered a weak inhibitor of CYP3A4 [24], but in vitro inhibition studies indicate that two of its metabolites, para-hydroxy-lactone and ortho-hydroxy-lactone, may be inhibitors of CYP3A4 [29]. Furthermore, at least one atorvastatin metabolite, ortho-atorvastatin lactone, appears to be an inhibitor of CYP3A4 [29]. In our study, atorvastatin was administered for 14 days, a sufficient amount of time to reach a steady state necessary for the maximal inhibition of hepatic CYP3A4. It is possible that such inhibition of CYP3A4 might affect the hepatic metabolism of colchicine to inactive metabolites, leading to an increase in colchicine plasma concentrations.
It should be noted that the potential for CYP3A4-related interactions between various statins and colchicine is likely to be variable, as different statins are metabolized by CYPS3A4 to different extents. For example, rosuvastatin is not metabolized by CYP3A4 at all [30], while simvastatin is primarily metabolized by this isoenzyme [31]. As such, our results cannot be applied to other statin/colchicine interactions.
Atorvastatin, like other statins, is a direct P-gp inhibitor [20, 28]. Median inhibitory concentration values of atorvastatin acid and atorvastatin lactone needed for P-gp inhibition are 30 and 5.2 μmol/L, respectively [20]. With repeated administration of atorvastatin 40 mg once daily, mean plasma C max values for atorvastatin acid and atorvastatin lactone were 22 and 7.7 μmol/L, respectively [28]. So, although plasma concentrations of atorvastatin acid may not be high enough to inhibit P-gp, atorvastatin lactone plasma concentrations are. Furthermore, although colchicine efflux is affected by hepatic and renal P-gp transport [15], colchicine also undergoes significant P-gp-mediated efflux into the gastrointestinal lumen [13, 14]. Local concentrations of atorvastatin in the gastrointestinal tract can be as high as 70–550 μmol/L, which may have a potential impact on intestinal P-gp [32]. As such, atorvastatin may primarily interfere with P-gp–mediated transport of colchicine in the intestinal lumen, allowing for reduced colchicine efflux and, therefore, higher absorption. Other drug–drug interaction studies between colchicine and inhibitors of CYP3A4 or P-gp indicate that P-gp inhibition has a greater influence on colchicine bioavailability than does CYP3A4 inhibition [23].
A limitation of this study could be the sample size, which was based on those used in prior atorvastatin and colchicine drug–drug interaction studies; no formal sample size calculation was performed. Therefore, it cannot be ruled out that in a larger number of subjects, myotoxic AEs would not have been observed. A second limitation of this study may be the high percentage of men enrolled (92 %). It is known that gender-related differences in the pharmacokinetics of atorvastatin exist. In fact, in women, atorvastatin C max values can be approximately 20 % higher and the AUC approximately 10 % lower [33]. Because only two women enrolled in our study, it is not possible to determine if the gender-based differences in atorvastatin pharmacokinetics would impact any colchicine–atorvastatin interaction.
The dose of colchicine chosen for this study, 0.6 mg, is the daily dose recommended for gout flare prophylaxis [5]. Per the FDA drug–drug interaction guidance, a single dose of the substrate is the most sensitive setting for evaluating drug interactions [24]. The selected dose regimen of atorvastatin was at the higher end of the recommended dose range (10–80 mg/day) to maximize the potential of an interaction. The duration of atorvastatin dosing (14 days) was selected to ensure adequate time to achieve a steady state and maximal pharmacologic effects on enzymes involved in colchicine metabolism. Our study was not designed to determine whether steady-state colchicine therapy increased plasma atorvastatin concentrations, nor did it investigate the myocyte accumulation of colchicine and/or statin. Since gout management guidelines recommend the extended use (≥6 months) of colchicine for gout flare prophylaxis during initial urate-lowering therapies [2], studies are needed to determine the potential long-term impact of atorvastatin on the tolerability and safety of long-term colchicine use and vice versa.
5 Conclusions
The results of this study show an increase in both peak (31 %) and total exposure (approximately 25 %) of colchicine following co-administration of a single dose of colchicine with steady-state atorvastatin, and the 90 % CIs for the ratios of the central values of C max, AUClast, and AUC∞ were outside the no-effect range of 80–125 %. However, rates of AEs were low with no clear differences between the regimens (colchicine alone or co-administered with atorvastatin) and no consistent changes in laboratory test values, vital signs, or physical findings were observed. While the safety results of this single-dose study suggest minimal clinical effect, additional investigations are warranted to explore if steady-state colchicine concentrations with steady-state atorvastatin concentrations could possibly exacerbate each other’s myopathic potential. As such, clinicians should monitor for colchicine-related toxicities during concomitant long-term administration of colchicine and atorvastatin. Caution is warranted during the co-administration of these agents, particularly in the elderly, in those with renal or hepatic dysfunction where the dose of colchicine may be appropriately reduced, or in polymedicated patients.
References
Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63:3136–41.
Khanna D, Khanna PP, Fitzgerald JD, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447–61.
Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am J Med. 2012;125(679–87):e1.
Primatesta P, Plana E, Rothenbacher D. Gout treatment and comorbidities: a retrospective cohort study in a large US managed care population. BMC Musculoskelet Disord. 2011;12:103.
Colcrys (colchicine, USP) tablets for oral use. Full prescribing information. Deerfield: Takeda Pharmaceuticals America, Inc.; 2012.
Tufan A, Dede DS, Cavus S, et al. Rhabdomyolysis in a patient treated with colchicine and atorvastatin. Ann Pharmacother. 2006;40:1466–9.
Sahin G, Korkmaz C, Yalcin AU. Which statin should be used together with colchicine? Clinical experience in three patients with nephrotic syndrome due to AA type amyloidosis. Rheumatol Int. 2008;28:289–91.
Hsu WC, Chen WH, Chang MT, et al. Colchicine-induced acute myopathy in a patient with concomitant use of simvastatin. Clin Neuropharmacol. 2002;25:266–8.
Justiniano M, Dold S, Espinoza LR. Rapid onset of muscle weakness (rhabdomyolysis) associated with the combined use of simvastatin and colchicine. J Clin Rheumatol. 2007;13:266–8.
Alayli G, Cengiz K, Canturk F, et al. Acute myopathy in a patient with concomitant use of pravastatin and colchicine. Ann Pharmacother. 2005;39:1358–61.
Atasoyu EM, Evrenkaya TR, Solmazgul E. Possible colchicine rhabdomyolysis in a fluvastatin-treated patient. Ann Pharmacother. 2005;39:1368–9.
Sarullo FM, Americo L, Di Franco A, et al. Rhabdomyolysis induced by co-administration of fluvastatin and colchicine. Monaldi Arch Chest Dis. 2010;74:147–9.
Niel E, Scherrmann JM. Colchicine today. Joint Bone Spine. 2006;73:672–8.
Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum. 2009;38:411–9.
Simkin PA, Gardner GC. Colchicine use in cyclosporine treated transplant recipients: how little is too much? J Rheumatol. 2000;27:1334–7.
Speeg KV, Maldonado AL, Liaci J, et al. Effect of cyclosporine on colchicine secretion by a liver canalicular transporter studied in vivo. Hepatology. 1992;15:899–903.
Speeg KV, Maldonado AL, Liaci J, et al. Effect of cyclosporine on colchicine secretion by the kidney multidrug transporter studied in vivo. J Pharmacol Exp Ther. 1992;261:50–5.
Chen YJ, Huang SM, Liu CY, et al. Hepatobiliary excretion and enterohepatic circulation of colchicine in rats. Int J Pharm. 2008;350:230–9.
Leighton JA, Bay MK, Maldonado AL, et al. The effect of liver dysfunction on colchicine pharmacokinetics in the rat. Hepatology. 1990;11:210–5.
Bogman K, Peyer AK, Torok M, et al. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol. 2001;132:1183–92.
Boyd RA, Stern RH, Stewart BH, et al. Atorvastatin coadministration may increase digoxin concentrations by inhibition of intestinal P-glycoprotein-mediated secretion. J Clin Pharmacol. 2000;40:91–8.
Choi DH, Shin WG, Choi JS. Drug interaction between oral atorvastatin and verapamil in healthy subjects: effects of atorvastatin on the pharmacokinetics of verapamil and norverapamil. Eur J Clin Pharmacol. 2008;64:445–9.
Terkeltaub RA, Furst DE, Digiacinto JL, et al. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 2011;63:2226–37.
Food and Drug Administration Guidance for Industry. Drug interaction studies: study design, data analysis, implication for dosing, and labeling recommendations. February 2012. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed 15 Aug 2013.
Baker SK, Goodwin S, Sur M, et al. Cytoskeletal myotoxicity from simvastatin and colchicine. Muscle Nerve. 2004;30:799–802.
Francis L, Bonilla E, Soforo E, et al. Fatal toxic myopathy attributed to propofol, methylprednisolone, and cyclosporine after prior exposure to colchicine and simvastatin. Clin Rheumatol. 2008;27:129–31.
Bouquie R, Deslandes G, Renaud C, et al. Colchicine-induced rhabdomyolysis in a heart/lung transplant patient with concurrent use of cyclosporin, pravastatin, and azithromycin. J Clin Rheumatol. 2011;17:28–30.
Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42:1141–60.
Lea AP, McTavish D. Atorvastatin: a review of its pharmacology and therapeutic potential in the management of hyperlipidaemias. Drugs. 1997;53:828–47.
Crestor (rosuvastatin calcium) tablets. Full prescribing information. Wilmington: AstraZeneca Pharmaceuticals LP; 2013.
Zocor (simvastatin) tablets. Full prescribing information. Whitehouse Station: Merck & Co., Inc.; 2012.
Wu X, Whitfield LR, Stewart BH. Atorvastatin transport in the Caco-2 cell model: contributions of P-glycoprotein and the proton-monocarboxylic acid co-transporter. Pharm Res. 2000;17:209–15.
LIPITOR® (atorvastatin calcium) tablets. Full prescribing information. New York: Pfizer, Inc.; 2012. http://www.lipitor.com/index.aspx. Accessed 8 Aug 2013.
Acknowledgments
The authors would like to acknowledge Robert Faulkner, PhD, Thomas Lauterio, PhD (Mutual Pharmaceutical Company, Inc.), Jennifer DiGiacinto, PhD (Salamandra LLC, Bethesda), Michael Mayer (Takeda Developmental Center Americas, Inc.), and Robert Terkeltaub, MD (San Diego VA Medical Center and UCSD, Division of Rheumatology Allergy, and Immunology) for their critical review and revisions of the manuscript for important intellectual content. Editorial assistance was provided by Deepa Mothey, PhD, and Meryl Gersh, PhD, of AlphaBioCom, LLC, and was supported by Takeda Pharmaceuticals International, Inc.
This study (ClinicalTrials.gov registered study number NCT00960323) was sponsored and completed by Mutual Pharmaceutical Company, Inc. prior to becoming a part of the Takeda Pharmaceuticals USA Inc. family of companies (Deerfield, IL, USA). Takeda Pharmaceuticals International, Inc. did not have an influence on study design, data collection, or analysis and interpretation of the data, and reviewed the content for medical accuracy and supported the decision to publish the manuscript. Matthew W. Davis and Suman Wason were employees of Mutual Pharmaceutical Company, Inc. at the time of study design, conduct, and analysis prior to Mutual Pharmaceuticals becoming a wholly owned subsidiary of Takeda Pharmaceutical Company Ltd.
Data from this study were presented in part in an abstract and poster at the American College of Cardiology 60th Annual Scientific Session and Expo, 2–5 April 2011, New Orleans, LA, USA.
Author information
Authors and Affiliations
Corresponding author
Additional information
ClinicalTrials.gov identifier: NCT00960323.
M. W. Davis and S. Wason were affiliated in Mutual Pharmaceutical Company, Inc., now a part of the Takeda Pharmaceuticals USA Inc. Family of Companies, Deerfield, IL, USA at the time of the study.
Rights and permissions
About this article
Cite this article
Davis, M.W., Wason, S. Effect of Steady-State Atorvastatin on the Pharmacokinetics of a Single Dose of Colchicine in Healthy Adults Under Fasted Conditions. Clin Drug Investig 34, 259–267 (2014). https://doi.org/10.1007/s40261-013-0168-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-013-0168-8