
Abstract
Dermatomyositis is a rare inflammatory disease with characteristic cutaneous findings and varying amounts of systemic involvement. Patients may present with skin disease alone, have concomitant muscle disease, or have extracutaneous manifestations such as pulmonary disease or an associated malignancy. Given such diverse presentations, dermatomyositis is both a diagnostic and therapeutic challenge. However, a prompt diagnosis is of utmost importance to institute adequate therapy and screen patients for an associated malignancy. Dermatologists should play a crucial role in the diagnosis and management of patients with dermatomyositis as cutaneous disease tends to be chronic, negatively impact quality of life, and be more recalcitrant to therapy. In this review, we discuss diagnosis, with a focus on myositis-specific antibodies and their associated phenotypes. We also review therapies available for this often refractory skin disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Myositis-specific autoantibodies are associated with characteristic clinical features and can alert physicians to potentially associated systemic manifestations. |
The skin disease of dermatomyositis can be particularly challenging to manage. |
Treatment must be personalized depending on patient comorbidities and preference, risk–benefit ratio, and the presence of any associated internal manifestations. |
1 Introduction
Dermatomyositis (DM) is an idiopathic multi-system inflammatory condition. Adult DM, which affects women more than men, remains a rare disease with an annual incidence of 1 per 100,000 persons, though incidence may be increasing [1,2,3,4]. While the exact pathogenesis of DM is still not fully elucidated, studies have shown abnormal and upregulated signaling through the interferon pathway [5, 6].
Classic DM (CDM) presents with pathognomonic cutaneous findings and progressive, symmetric proximal muscle weakness. Cutaneous disease precedes the appearance of myositis by 3–6 months in 30–50% of patients, while 10% of patients present with muscle symptoms prior to the development of skin findings [7, 8]. There is a subset of patients with DM (approximately 20%) who have a skin-predominant phenotype and are classified as clinically amyopathic DM (CADM) [1, 4, 9, 10]. Of note, the diagnosis of CADM is provisional at 6 months and confirmed at 2 years [10], and encompasses both amyopathic DM and hypomyopathic DM. In a large review of 291 patients with CADM, 70% had amyopathic DM and 13% had hypomyopathic DM [11]. Although both of these subtypes have no clinical evidence of muscle involvement, there is subclinical evidence of muscle involvement demonstrated on laboratory, electrophysiologic, or radiologic evaluations in the hypomyopathic variant [1, 9, 12]. Both patients with CDM and patients with CADM have an elevated risk of developing interstitial lung disease and occult malignancy [11, 13, 14]. Post-myopathic DM refers to a subset of patients who have resolution of their muscle involvement with therapy, but have persistent cutaneous disease [8, 11].
Dermatologists should play an integral role in the diagnosis and management of patients with DM as cutaneous involvement is evident in all DM subtypes, often persists after successful treatment of muscle disease, and can greatly impact quality of life [8, 15]. The course of cutaneous disease tends to be chronic and prolonged. A prospective cohort of 74 patients with DM receiving various systemic regimens found that only 38% had achieved remission of skin disease in a 3-year follow-up period [16]. With regard to CADM specifically, the role of dermatologists is crucial as many providers have difficulty recognizing DM in the absence of muscle involvement, which often leads to misdiagnosis and contributes to delays in treatment and an appropriate initial workup [11, 13, 17]. Da Silva et al. found the median delay to correct diagnosis was 17.1 months in patients with CADM, which was significantly higher than patients with CDM (12.2 months) [17]. This delay is clinically relevant, particularly with regard to adequately screening patients for malignancy given that the risk is highest in the first 2 years after symptom onset [14, 18, 19].
2 Diagnosis
Given DM’s protean manifestations, a detailed physical exam is at the cornerstone of making the correct diagnosis. The cutaneous features of DM include the pathognomonic findings of Gottron’s papules (pink-violaceous papules on the dorsal hands, with a predilection for the skin overlying the metacarpophalangeal and interphalangeal joints) and heliotrope eruption (pink-violaceous erythema involving the upper eyelids, at times accompanied with edema) [7]. Other characteristic findings are Gottron’s sign (macular erythema or pink-violaceous papules overlying joints), photodistributed pink-violaceous erythema or poikiloderma of the upper back (“shawl” sign) and anterior neck and upper chest (“V” sign), and nailfold abnormalities (periungual erythema, dilated capillary nail bed loops with alternating areas of drop out, and cuticular hypertrophy) [3, 7]. Patients often also have midfacial erythema involving the nasolabial folds, unlike the malar erythema of acute cutaneous lupus erythematosus, which spares the nasolabial folds. Additional cutaneous findings of DM include pink-violaceous scaly erythema or poikiloderma of lateral thighs (“holster sign”) and scalp involvement (erythema and psoriasiform scaling, often with associated non-scarring alopecia), amongst others. Of note, scalp involvement can be extremely symptomatic and scalp dysesthesia may occur in patients without evidence of an eruption [7].
Expert clinicians can usually arrive at a correct diagnosis of DM with a physical exam alone. However, a skin biopsy may be helpful if findings on exam are subtle or atypical. Skin biopsy demonstrates a vacuolar interface dermatitis with dermal mucin deposition. Physicians should be aware that these findings are also seen in lupus erythematosus. Hence, these two entities are difficult to distinguish on histology alone [20].
When evaluating muscle disease, a detailed history with pointed questions (difficulty combing hair, getting out of a seated position, difficulty swallowing, change in voice) and strength testing of muscle groups should be performed at each clinic visit. Muscle enzymes should be trended periodically for the first 2 years. When the clinical diagnosis is in question, or when the patient has normal muscle enzymes in the presence of clinical weakness, additional investigations may be warranted, including magnetic resonance imaging or ultrasound of proximal muscles, electromyography, or muscle biopsy [3, 21].
Patients should be screened regularly for pulmonary symptoms given the prevalence of interstitial lung disease (ILD) in 5–35% of patients with DM [22,23,24,25,26,27]. A thorough review of symptoms (cough, shortness of breath, dyspnea on exertion) is necessary at each clinic visit. Evaluation with pulmonary function tests with a diffusion capacity for carbon monoxide are warranted at baseline [28, 29]. If the pulmonary function tests demonstrate abnormal findings, a high-resolution chest computed tomography scan with an ILD protocol should be performed. Pulmonary function tests should be repeated every 3–12 months, depending on the initial findings and the risk of ILD in a particular patient (e.g., high-risk subtype) [30].
Myositis-specific autoantibodies (MSAs) are found only in patients with idiopathic inflammatory myopathies (DM, polymyositis, inclusion body myositis, and necrotizing myopathies). In recent years, more studies have focused on identifying MSAs in DM and describing their associated phenotype. The majority of patients with DM only have one MSA [31, 32], and only approximately 20% of patients with DM have a known MSA [33]. The gold standard for detection of MSAs is the immunoprecipitation assay, which is not widely available and lacks standardization, currently limiting the practical use of these antibodies [26].
It is important for clinicians to recognize the specific MSA-associated phenotypes as these can help with prognostication, alerting the clinician of systemic manifestations that are more likely in a patient. Despite the usefulness of the MSA-associated phenotypes, there is a considerable overlap of clinical features amongst some of the MSA groups [31].
-
Mi-2 antibody: The prevalence of Mi-2 antibodies in adult patients with DM ranges from 2 to 38% [33, 34]. Patients present with classic skin findings and myositis and have a decreased incidence of both ILD and malignancy compared with other patients with DM [33,34,35,36]. Overall, patients have a favorable prognosis and respond well to therapy [33, 37]. Longitudinal clinical monitoring is warranted as patients often have recurrence of disease with cessation of therapy [33].
-
Anti-SAE1/2 antibody: The prevalence of anti-SAE1/2 autoantibodies ranges from 1 to 10% [26, 31, 32, 38]. These patients tend to have classic cutaneous findings, myositis, and dysphagia [22, 25, 39,40,41]. Several cohorts have reported a novel diffuse red-violaceous exanthem, which may ulcerate [22, 39,40,41,42]. An increased risk of mild ILD and malignancy in patients with anti-SAE1/2 antibodies has been reported, but only in small cohorts to date, warranting further investigation [22, 25, 40, 41]. Interestingly, four patients with anti-SAE antibodies in Asian cohorts have had pulmonary arterial hypertension that could not be attributed simply to the degree of ILD [40, 41]. This also warrants additional investigation.
-
Anti-aminoacyl-transfer RNA synthetase (ARS) antibodies: Eight anti-ARS autoantibodies (anti-Jo-1, anti-OJ, anti-EJ, anti-KS, anti-Zo, anti-Ha/YRS, anti-PL-12, and anti-PL-1) have been associated with the anti-synthetase syndrome. Anti-Jo-1is the most common, with prevalence as high as 20% [31, 43]. The clinical presentation of anti-synthetase syndrome is quite heterogeneous and varies by anti-ARS antibody [44]. The “classic” clinical triad consists of ILD, myositis, and arthritis. Other associated features include fever, Raynaud’s phenomenon, and mechanic’s hands (dry, fissured, hyperkeratotic skin on the lateral and palmar hands and fingers). A Japanese cohort noted that the majority of their patients with the anti-ARS antibody who initially presented with only myositis later developed ILD, emphasizing that longitudinal monitoring is necessary [43]. For patients who develop ILD, the overall prognosis is favorable, with a 5-year-survival rate of 96% [45].
-
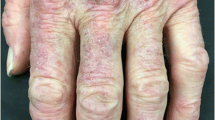
Anti-melanoma differentiation antigen 5 (MDA-5) antibody: These autoantibodies, which are more prevalent in Asian (11–57%) compared with Caucasian (0–13%) cohorts [31, 46], convey an increased risk of developing ILD, including a rapidly progressive variant with high mortality [26, 27, 47]. One group reported a 90-day survival rate of only 66% for MDA-5-positive patients with ILD. In contrast, patients with ILD and anti-ARS antibodies had a survival rate of 100% [45]. Patients with MDA-5 DM have a higher prevalence of amyopathic disease (50–77%), fevers, and inflammatory arthritis [24, 27, 31, 33, 48]. Fiorentino et al. described a characteristic cutaneous phenotype in patients with MDA-5-positive DM, including painful erythematous palmar papules and macules (Fig. 1), cutaneous ulcerations of the digital pulp, nailfolds, and over the Gottron’s papules and sign, oral erosions, prominent non-scarring alopecia, and mechanic’s hands [24, 27]. These patients tend to have severe skin disease that is less likely to achieve clinical remission despite systemic therapy [16].
Fig. 1 
Patient with MDA-5 dermatomyositis with tender erythematous papules and macules on palms and interphalangeal creases
-
Anti-TIF-1γ antibody: These autoantibodies are more prevalent in Caucasian (41%) compared with Asian (17%) cohorts [32, 49,50,51,52]. Although there is a well-established association of malignancy in patients with anti-TIF-1γ antibodies, the risk may be influenced by several factors including male sex, older age, and smoking [26, 33, 49, 53,54,55]. Ethnicity may also be a factor, but more studies are necessary to validate this observation [49]. Patients with anti-TIF-1γ antibodies tend to have clinical evidence of myositis and lower prevalence of ILD, Raynaud’s phenomenon, and arthralgias [33, 49]. Patients tend to have severe cutaneous disease, albeit with a decreased risk of calcinosis cutis [49, 53]. In addition to the classic photodistributed eruptions, these patients may also have asymptomatic hyperkeratotic papules on the palms, psoriasiform lesions (Fig. 2), hyperkeratotic Gottron’s papules, red-on-white lesions (hypopigmented patches admixed with focal, often follicular, telangiectatic macules), and an ovoid palatal patch [49, 56].
Fig. 2 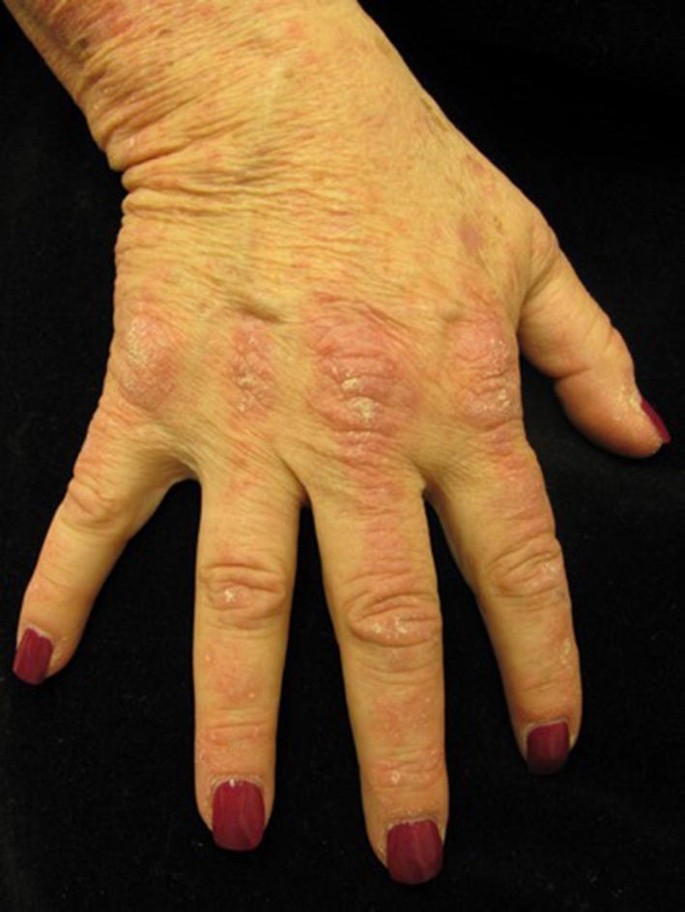
Patient with TIF-1γ dermatomyositis with Gottron’s papules with psoriasiform scale on the metacarpophalangeal, proximal, and distal interphalangeal joints
-
Anti-NXP2 (MJ) antibody: The prevalence of anti-NXP2 autoantibodies in adult patients with DM ranges from 2 to 30% [57]. Patients with anti-NXP2 antibodies often present with severe recurrent myalgias, both proximal and distal weakness, and severe dysphagia [57, 58]. Although patients have milder cutaneous findings, unique to their presentation is increased peripheral edema and calcinosis cutis [57,58,59]. Patients with NXP2-positive DM are also at an increased risk of developing malignancy, while the prevalence of ILD in this population is decreased compared with other patients with DM [31, 39, 58, 60, 61].


Importantly, MSAs are currently used to phenotype and stratify patients with DM rather than to make the diagnosis, given that they are only present in approximately 20% of patients with DM. When present, MSAs can serve to help confirm a diagnosis of DM. Presently, there are insufficient data to make formal guidelines regarding how to use MSAs to guide clinical management. However, with data from larger prospective cohorts and a standardized method of detection, there is potential to use MSAs to optimize management strategies in patients with DM.
Once DM is diagnosed, a thorough screening for internal malignancy is warranted. Multiple studies have substantiated that adult patients with DM have an elevated risk of malignancy, although the frequency (9–42%) and type of malignancy vary greatly in different studies [14, 18, 61,62,63,64,65]. A retrospective study of a US cohort of 400 patients with DM reported the risk of malignancy to be 12%, with no significant difference between CDM and CADM subtypes [14]. A meta-analysis found that several factors (older age, male sex, cutaneous necrosis, cutaneous vasculitis, dysphagia, elevated Erythrocyte sedimentation rate (ESR), and rapid onset of myositis) were associated with an increased malignancy risk and also found the presence of interstitial lung disease, arthralgia, and Raynaud’s phenomenon to be protective [66]. As noted above, both anti-TIF-1γ and anti-NXP2 antibodies convey an increased risk of malignancy [66, 67]. Further studies are needed to determine whether these clinical factors and antibodies can be used to stratify patients with DM based on their malignancy risk, but at present, malignancy screening is recommended for all adult patients diagnosed with DM [14].
3 Management
Each patient with DM requires an individualized therapeutic plan that takes into account the cutaneous disease severity, presence of concomitant muscle disease, systemic involvement, other comorbidities, including underlying malignancy, and the overall impact of disease on a patient’s quality of life. With regard to skin disease specifically, the treatment goal is to obtain control of cutaneous disease with the safest combination of therapeutics.
The treatment of skin disease in DM can be particularly challenging, given that cutaneous DM is often more recalcitrant to treatment than the muscle involvement in DM [2, 68]. Despite this challenge, clinicians should strive to optimize the treatment of cutaneous disease as the associated pruritus, photosensitivity, and appearance of skin lesions can significantly impact quality of life [12, 15, 69].
The majority of data for the treatment of cutaneous DM comes from expert opinion, case series, retrospective reviews, and open-label studies. There is a paucity of randomized controlled trials. Additionally, interpretation of the available literature is difficult owing to several factors in existing studies including: pooling of various inflammatory myopathies (i.e., polymyositis and DM), lack of use of a standardized measure (i.e., Cutaneous Dermatomyositis Disease Area and Severity Index [CDASI] or Dermatomyositis Skin Severity Index [DSSI]) to assess cutaneous response to therapy, primary focus on resolution of myositis, and concomitant administration of immunosuppressive therapies for muscle disease [70,71,72]. Despite these limitations, several management principles can be established.
3.1 Initial Management
First-line therapy should include aggressive photoprotection, antipruritic agents, and topical anti-inflammatory medications (corticosteroids and calcineurin inhibitors). A minority of patients can achieve remission of their cutaneous disease with these interventions alone. In the vast majority of patients with DM, these therapies should be used as adjunctive therapies to systemic agents given the refractory nature of DM skin disease.
3.2 Photoprotection
It is well established that ultraviolet light can induce or flare cutaneous DM; therefore, strict photoprotection is necessary [12, 68, 73, 74]. Patients should be counseled on the need to practice sun protection on a year-round basis, not only during the summer months. A broad-spectrum sunscreen (with a sun protective factor of at least 50) should be used daily and reapplied every 2 h [2, 68, 75]. Sun avoidance, wide-brimmed hats, and sun-protective clothing should also be strongly encouraged. Given the level of photoprotection recommended, clinicians should consider the assessment of vitamin D levels and provide supplementation if needed.
3.3 Antipruritic Agents
Pruritus is often a debilitating feature of DM that can negatively impact a patient’s quality of life, alter sleep patterns, and interfere with activities of daily living [15, 69, 76]. Aggressive management with a combination of proper skin care with bland emollients to minimize xerosis, oral antihistamines, and other anti-pruritic agents such as amitriptyline or gabapentin may be used [68, 74]. Immunosuppressive therapy may be warranted for intractable pruritus, even in the case of what may appear to be mild skin disease [74]. Elevated levels of interleukin-31 have been implicated in DM-associated itch, and lenabasum (JBT-101) is a non-psychoactive cannabinoid that suppresses interleukin-31 levels [77]. In a phase II study and open-label extension of patients with DM with skin-predominant refractory disease, lenabasum-treated subjects had a clinically significant decrease in CDASI activity scores and improvement in multiple patient-reported outcomes [78, 79].
3.4 Topical Therapy: Corticosteroids and Calcineurin Inhibitors
Topical therapy can serve as adjunctive treatment, but very rarely controls cutaneous disease as monotherapy [68]. Topical corticosteroids can be used to decrease erythema and pruritus. The strength and vehicle selected for topical corticosteroids depend on the site of application and patient preference. Stronger topical corticosteroids (group I and II) are generally reserved for areas with thicker skin such as the scalp, hands, and extensor surfaces, while lower potency topical corticosteroids (group VI and VII) can be used on thinner areas more prone to atrophy, such as the face. Use of high-potency corticosteroids under occlusion can increase efficacy, particularly for the dorsal hands [8].
Topical calcineurin inhibitors include tacrolimus and pimecrolimus. While data are mixed regarding their efficacy for cutaneous DM, most studies have found a positive effect [80,81,82,83,84,85]. An advantage of topical calcineurin inhibitors is that they can be used on areas with thinner skin without the risk of atrophy. Patients should be warned regarding the local side effect of burning with initial application, although these symptoms usually abate with repeated use [86, 87].
4 Systemic Therapy
As noted above, most patients with cutaneous DM require systemic medications. The choice of systemic agent should be tailored for each patient and is dependent on the presence of other manifestations of DM, predominantly myositis or lung involvement. Here, we focus on the therapies used most commonly in clinical practice for cutaneous DM in adult patients.
4.1 Antimalarials
For many years, antimalarials [hydroxychloroquine (HCQ), chloroquine (CQ), and quinacrine] have been the preferred initial treatment for cutaneous DM given their long history of use and overall tolerability. Although their exact mechanism of action is unknown, antimalarials have an anti-inflammatory effect and are photoprotective [88]. In Europe, CQ is favored as it is thought to be more effective; however, in the USA, HCQ is preferred owing to the greater risk of irreversible retinopathy associated with CQ [8].
Multiple case series, retrospective reviews, and open-label studies have found antimalarials to be beneficial for skin manifestations of DM, but not for myositis [88,89,90,91,92,93]. Although one series of seven patients demonstrated a complete clinical response in 43% of patients with cutaneous DM [89], a more recent retrospective review of 115 patients at four tertiary care centers demonstrated that only 11% of patients with cutaneous DM responded adequately to antimalarial therapy without requiring escalation to additional agents [94]. Furthermore, up to one-third of patients with DM develop a cutaneous drug reaction, typically a morbilliform eruption, with initiation of HCQ [94, 95]. Some patients that develop a drug reaction to HCQ may progress to tolerating CQ [96]. One retrospective cohort study found that patients with anti-SAE-1/2 autoantibodies were at a higher risk of developing a drug eruption to HCQ, while no patients with anti-MDA-5 autoantibodies had a drug reaction [52].
If cutaneous disease is not adequately controlled with HCQ, combining it with quinacrine (100 mg daily), owing to a possible synergistic effect, or switching to CQ are options [8, 75]. A small retrospective study found that 7/17 patients (41%) had near clearance of cutaneous symptoms with use of antimalarial therapy alone (three were controlled with HCQ alone and four required a combination of either HCQ and quinacrine or CQ and quinacrine) [97]. Hydroxychloroquine and CQ should never be combined because of the additive ocular toxicity. Of note, for patients with severe cutaneous disease, the authors favor adding methotrexate (MTX) to HCQ, rather than combining antimalarials or switching to CQ. In our experience, this provides a more robust response in patients with severe skin disease.
Based on recently updated guidelines from the American Academy of Ophthalmology, the total dose of HCQ should be less than or equal to 5 mg/kg/day based on actual body weight, while the total dose of CQ should be less than or equal to 2.3 mg/kg/day based on actual body weight [98]. The response to antimalarials is usually not evident until 6–8 weeks after initiation of therapy [8], or even up to 12 weeks. Hydroxychloroquine and CQ are usually well tolerated but can cause a gastrointestinal upset, hypersensitivity reactions, and blue-gray dyspigmentation [68, 88]. Rarer adverse effects include: transaminitis, bone marrow toxicity, neuropathy, myopathy, and cardiomyopathy [12, 88, 99]. Their most feared side effect is irreversible retinopathy; thus, patients taking HCQ or CQ need a baseline fundus examination and regular follow-up with ophthalmology [98]. Quinacrine does not cause ocular toxicity, but can cause a reversible yellow discoloration of the skin and, rarely, aplastic anemia [68]. In the USA, quinacrine is only available at compounding pharmacies.
4.2 Methotrexate
Methotrexate is an antimetabolite with both anti-proliferative (through inhibition of dihydrofolate reductase, which ultimately leads to inhibition of cell division) and anti-inflammatory (through inhibition of 5-aminoimidazole-4-carboxamide ribonucleotide transformylase) properties. Inhibition of 5-aminoimidazole-4-carboxamide ribonucleotide transformylase leads to increased levels of adenosine, which is a purine with potent anti-inflammatory effects [100].
Methotrexate is often considered a first-line systemic therapy for cutaneous DM, particularly in those patients who are recalcitrant to or intolerant of antimalarial therapy. Importantly, it is effective for both cutaneous and muscle disease, making it an excellent corticosteroid-sparing agent for CDM [101]. It also may help in patients with associated joint symptoms. The majority of data supporting the use of MTX for cutaneous DM originates from case series and retrospective reviews [102,103,104,105,106,107]. Most recently, Hornung et al. conducted a retrospective analysis of 11 patients with systemic corticosteroid-resistant cutaneous DM and found a 73% response rate to MTX as evident by a mean decrease in the CDASI score from 14.1 to 5.5 (p < 0.1) [107].
Although the side-effect profile (including nausea, fatigue, malaise, hepatotoxicity, bone marrow toxicity, pneumonitis, mucositis, teratogenicity, and reversible oligospermia) of MTX is well established, the incidence of adverse events (AEs) in patients with DM taking MTX is quite variable in the literature. For example, one series of 13 patients with DM had no reported AEs, but in a different series of ten patients with DM, 70% had AEs attributed to MTX and 50% required drug discontinuation [104, 105]. This can be at least partially explained by the lack of standardization pertaining to MTX administration (route, dosing schedule), different folic acid supplementation practices, and the comorbidities of patients included [106]. Thus, patients must be carefully selected, and several factors should be assessed when considering MTX, including the presence of metabolic syndrome, alcohol consumption, non-alcoholic fatty liver disease, use of hepatotoxic medications, liver and renal function, concomitant pulmonary disease, and family planning. Because MTX can rarely cause pulmonary toxicity, its use is typically avoided in patients with DM with lung involvement [108].
At the authors’ institution, MTX is considered the first-line systemic agent considered for patients with CDM, patients with CADM intolerant of or recalcitrant to antimalarials, and patients with CADM with severe cutaneous disease at initial presentation. Patients are carefully screened and baseline laboratory studies (complete blood count with differential, blood urea nitrogen/creatinine, liver function tests, hepatitis serologies) are obtained. Methotrexate is often given in doses of 25 mg weekly for DM, with folic acid supplementation of 1 mg daily. At the authors’ institutions, MTX is started at a dose of 10 mg/week, with follow-up laboratory studies drawn at 2 weeks. If results are normal, the dose is typically escalated to 25 mg/week. As absorption of MTX is decreased at doses higher than 15 mg/week, we often split the dose (12.5 mg twice daily) [109]. Subsequent laboratory monitoring is conducted in 4–6 weeks and then every 2–3 months if the MTX dose remains stable. Patients are made aware that similar to antimalarials, MTX takes at least 6–12 weeks of continuous therapy to have a noticeable effect [74].
4.3 Systemic Corticosteroids
Systemic corticosteroids (SCS) have remained the cornerstone of initial therapy for patients with DM with active muscle disease [101]. Prednisone is typically started at doses of 0.5–1 mg/kg/day, but if the muscle involvement is severe or life threatening, intravenous methylprednisolone may be necessary [110, 111]. Corticosteroids are maintained at higher doses until muscle disease is quiescent and then are slowly tapered over several months. Some dermatologists use corticosteroids as initial treatment for cutaneous disease when it is severe, as a bridge until the effect of other systemic medication is evident [8, 96].
The myriad of AEs associated with prolonged SCS use are well established. Additionally, cutaneous and muscle disease tend to have a discordant response to therapy and skin disease is often more recalcitrant [75, 93]. While SCS may be necessary for DM-associated lung or muscle disease, their efficacy in cutaneous disease is more variable [12, 75, 112]. Therefore, the authors do not routinely use SCS as therapy for cutaneous disease unless there are concomitant extracutaneous DM manifestations that require their use.
4.4 Mycophenolate Mofetil
Mycophenolate mofetil (MMF) is a lymphocyte-selective immunosuppressive agent that inhibits inosine monophosphate dehydrogenase, an enzyme necessary for de novo purine synthesis. It exerts its immunosuppressive properties mainly through its potent cytostatic effect on lymphocytes, but also suppresses antibody formation and inhibits the recruitment of leukocytes into areas of inflammation [113, 114].
Mycophenolate mofetil is an effective agent for cutaneous disease, myositis, and DM-associated interstitial lung disease [113, 115,116,117,118,119,120,121,122]. The evidence for its efficacy in cutaneous disease comes from case series and uncontrolled studies. In 2006, Edge et al. conducted a review of 12 patients with DM with either refractory cutaneous disease or intolerance to more traditional agents, who were treated with MMF (dose range 1–4 g/day), and reported improvement in 83% of patients within 4–8 weeks [117]. A more recent prospective cohort study of 74 patients with moderate-to-severe cutaneous disease (CDASI activity score ≥ 12) found that treatment with MMF was significantly associated with achieving clinical remission [16]. The majority of patients who achieved clinical remission with MMF were treated with higher doses (3 g daily) [16]. Of note, in patients who have pulmonary involvement at presentation or are at increased risk (i.e., positive for the MDA-5 antibody or anti-synthetase syndrome) of developing DM-associated lung disease, MMF is the preferred first-line agent as it has been shown to allow for improvement in pulmonary function and have a corticosteroid-sparing effect [28, 123].
Mycophenolate mofetil is generally well tolerated. The most common AE is dose-dependent gastrointestinal distress (most commonly nausea, abdominal pain, diarrhea, and vomiting) [113]. Additionally, genitourinary symptoms may occur more commonly during the first year of therapy. It is teratogenic and can cause reversible cytopenias, and, as with any immunosuppressive agent, there is an increased risk of infection. There is a potential increased risk of malignancy with MMF, albeit the majority of malignancies reported with its use have been in the transplant population [113, 118].
Prior to starting MMF, required baseline laboratory studies include complete blood count with differential, blood urea nitrogen/creatinine, liver function tests, hepatitis serologies, and tuberculosis screening. We start MMF at 500 mg twice daily and recheck laboratory studies in 2 weeks. The dose is then titrated up to 1 g twice daily and in many patients subsequently increased to 1.5 g twice daily. Therapeutic effect from MMF is not seen until 6–12 weeks of therapy.
4.5 Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIg) is derived from pooled plasma from numerous donors. Although not entirely understood, proposed mechanisms of action in DM are neutralization of autoantibodies, downregulation of proinflammatory cytokines, binding of complement, and decreased formation and deposition of the membrane attack complex [124].
Intravenous immunoglobulin is effective for both refractory cutaneous disease and myositis. A double-blind placebo-controlled trial of 15 patients with DM refractory to various immunosuppressive agents showed significant improvements in muscle strength and neuromuscular symptoms in 9/12 (75%) patients. Additionally, 8/12 (67%) patients had marked improvement in their cutaneous disease as assessed by clinical photographs. Improvement was evident 15 days after the first infusion but peaked between the second and third month [125].
In a retrospective study, IVIg was added to the treatment regimen of 13 patients with DM with refractory cutaneous disease. All patients included were receiving antimalarial therapy, and 11/13 patients were taking at least one immunosuppressive agent. All patients had improvement, and a complete clinical response was seen in eight (62%) patients. Notably, IVIg had a corticosteroid-sparing effect and allowed for discontinuation of immunosuppressive agents in eight patients [126]. In another retrospective study of 27 patients with refractory cutaneous DM, IVIg was beneficial in 85% of the patients [127].
Intravenous immunoglobulin is typically well tolerated, with the most common AE being headache. Other rarer AEs include hypersensitivity reactions, aseptic meningitis, renal failure, myocardial infarction, and thrombosis. Given its expense, the authors typically reserve IVIg for patients with refractory cutaneous DM who have not responded to or are intolerant of first-line agents. Given its efficacy, however, we traditionally move quickly to IVIg for patients with very severe disease with poor quality of life as a result of their cutaneous DM. We use it both as monotherapy and as an adjunctive agent. Patients are usually treated with 2 g/kg of IVIg divided over 2 consecutive days every 4 weeks. Once clinical remission has been achieved, we typically increase the interval between treatments to every 5 weeks, then every 6 weeks, etc.
4.6 Rituximab
Rituximab is a chimeric monoclonal antibody that targets the CD20 antigen protein on B cells. It is an effective treatment for the extracutaneous manifestations of DM, particularly DM-associated interstitial lung disease and refractory myositis [28, 128,129,130]. For cutaneous disease, however, studies thus far have shown conflicting results.
In an open-label pilot study of seven patients with DM who received rituximab (RTX) [100 or 375 mg/m2 weekly for 4 weeks], all had improvement in strength and three patients with impaired pulmonary function at baseline had improvement in their forced vital capacity. At the beginning of the study, cutaneous disease, albeit with a limited description and listed only as “rash”, was documented in five patients and improved with treatment. Interpreting these results is challenging as a validated skin outcome measure was not utilized, and it is unclear what constituted improvement. Additionally, hair regrowth was noted in two patients with alopecia secondary to DM. Patients began relapsing around 24–36 weeks, which coincides with a return of B cells [131].
In contrast, an open-label study examined the effect of RTX (two 1-g doses separated by 2 weeks) added to the regimen of eight patients with DM with moderate-to-severe cutaneous disease, assessed by baseline DSSI scores. At week 24, three patients had achieved partial remission, which was defined as at least a 50% reduction in muscle strength deficit. However, the mean percentage of change in DSSI at week 24 was only 9.5%, which was not statistically significant. When evaluating specific cutaneous features through photographs, periungual telangiectasias and Gottron’s papules remain unchanged. The heliotrope eruption remained unchanged in six subjects, worsened in one, and improved in one. Poikiloderma was present in six subjects and either remained unchanged or worsened in three subjects [132]. In sum, the authors concluded that RTX could be useful in the treatment of muscle disease but has a minimal effect on skin disease.
A large prospective, multi-center, randomized, double-blind, placebo-controlled trial (RIM trial) of 200 patients with myositis, including 76 with adult DM and 48 with juvenile DM, who were refractory to SCS in addition to at least three other immunosuppressive agents, did not reach its primary outcome [128]. This may have been because of the trial design, given that all patients received RTX (either at weeks 0 and 1 or at weeks 8 and 9) in a randomized placebo-phase design. Despite not reaching its primary outcomes, a majority of patients in the study did experience improved muscle disease and a corticosteroid-sparing effect [128]. While the original trial did not assess the effect of RTX on cutaneous disease, a recent post hoc analysis on the RIM trial data found a beneficial effect. Lack of use of a validated skin outcomes measure and assessment of skin disease by non-dermatologists make it difficult to draw definitive conclusions [38]. At this time, given the limited evidence of efficacy for cutaneous DM specifically, the authors rarely use RTX as treatment for DM skin disease, and generally reserve it for our patients with refractory myositis or DM-associated pulmonary disease.
4.7 Janus Kinase Inhibitors
The Janus kinase (JAK)-signal transducer and activator of transcription (STAT) is an intracellular signaling pathway utilized by cytokines (including interleukins and interferons) and other molecules to transmit signals from the cell membrane to the nucleus. In recent years, its role in many inflammatory dermatoses has been better elucidated, and JAK inhibitors have been used successfully to treat various dermatologic conditions [133, 134].
In 2014, Hornung et al. reported a case of an elderly woman with CDM refractory to SCS, IVIg, MMF, and azathioprine, who received ruxolitinib, a JAK 1/2 inhibitor, for the treatment of post-polycythemia vera myelofibrosis. Her muscle strength improved, and cutaneous disease resolved completely while taking ruxolitinib [135].
In a subsequent series, one patient with CADM and two patients with CDM with refractory cutaneous disease were treated with tofacitinib, a JAK 1/3 inhibitor, 5 or 10 mg twice daily. All patients had improvement by week 4, as evidenced by a decrease in their CDASI activity score, and all reported a decrease in pruritus. The two subjects with CDM also reported improvement in strength and fatigue [136].
In the authors’ clinical experience, JAK inhibition can be an effective option for refractory cutaneous DM. Adverse events include an increased risk of infections, particularly herpes virus reactivation, gastrointestinal symptoms, laboratory abnormalities (dose-dependent increase in creatine phosphokinase, total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol; dose-dependent decrease in hemoglobin and neutrophil counts), and a potential increased risk of malignancy [133]. More recently, an increased risk of pulmonary embolism and overall mortality has been reported in a study of tofacitinib 10 mg twice daily in patients with rheumatoid arthritis [137]. It is unclear whether this association will be found in patients with other autoimmune conditions taking tofacitinib at 10 mg twice daily.
4.8 Other Therapies
Immunosuppressive agents such as cyclophosphamide, tacrolimus, sirolimus, cyclosporine, azathioprine, and chlorambucil have been used in treatment of refractory myositis and DM-associated lung disease [138,139,140,141,142,143,144,145,146,147,148]. However, evidence for efficacy in the management of cutaneous disease is limited and in the form of case reports and series [143, 149,150,151,152,153].
A few case reports have demonstrated an improvement in cutaneous DM with dapsone, leflunomide, and thalidomide [91, 154,155,156,157,158,159,160,161,162]. In a series of two patients with refractory cutaneous DM, dapsone was added to their regimen and both had a rapid response. Additionally, cutaneous disease flared when dapsone was stopped and improved with re-initiation [156]. Thalidomide can be effective in recalcitrant patients, but its use may be limited by the development of peripheral neuropathy [161].
The evidence for tumor necrosis factor-α antagonists is contradictory regarding their benefit in cutaneous and muscle disease [163,164,165,166,167]. Most importantly, several reports note inciting or worsening of DM, both skin and muscle disease, with their use [168,169,170,171,172,173,174,175,176,177,178,179,180,181,182]. For this reason, myositis experts typically consider anti-tumor necrosis factor therapies contraindicated in patients with DM.
5 Conclusions
Dermatomyositis is a rare idiopathic inflammatory disease with diverse presentations that can have varying degrees of cutaneous and systemic involvement. This heterogeneity in phenotype makes DM both a diagnostic and therapeutic challenge. Diagnosis relies heavily on a comprehensive physical exam. Dermatologists should be aware of specific MSA-associated phenotypes that can help them anticipate the most likely systemic associations in a particular patient. Overall, cutaneous DM tends to be chronic, debilitating, and often recalcitrant to therapy. Although treatment may be challenging, dermatologists should play an active role in the management of patients with cutaneous DM. Randomized controlled trials, which use validated skin outcomes measures, are needed to develop an evidence-based treatment algorithm for this frequently refractory skin disease.
References
Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146(1):26–30. https://doi.org/10.1001/archdermatol.2009.328.
Strowd LC, Jorizzo JL. Review of dermatomyositis: establishing the diagnosis and treatment algorithm. J Dermatol Treat. 2013;24(6):418–21. https://doi.org/10.3109/09546634.2012.697540.
Iaccarino L, Ghirardello A, Bettio S, Zen M, Gatto M, Punzi L, et al. The clinical features, diagnosis and classification of dermatomyositis. J Autoimmun. 2014;48–49:122–7. https://doi.org/10.1016/j.jaut.2013.11.005.
Galimberti F, Li Y, Fernandez AP. Clinically amyopathic dermatomyositis: clinical features, response to medications and malignancy-associated risk factors in a specific tertiary-care-centre cohort. Br J Dermatol. 2016;174(1):158–64. https://doi.org/10.1111/bjd.14227.
Kao L, Chung L, Fiorentino DF. Pathogenesis of dermatomyositis: role of cytokines and interferon. Curr Rheumatol Rep. 2011;13(3):225–32. https://doi.org/10.1007/s11926-011-0166-x.
Huard C, Gulla SV, Bennett DV, Coyle AJ, Vleugels RA, Greenberg SA. Correlation of cutaneous disease activity with type 1 interferon gene signature and interferon beta in dermatomyositis. Br J Dermatol. 2017;176(5):1224–30. https://doi.org/10.1111/bjd.15006.
Callen JP. Cutaneous manifestations of dermatomyositis and their management. Curr Rheumatol Rep. 2010;12(3):192–7. https://doi.org/10.1007/s11926-010-0100-7.
Sontheimer RD. The management of dermatomyositis: current treatment options. Expert Opin Pharmacother. 2004;5(5):1083–99. https://doi.org/10.1517/14656566.5.5.1083.
Sontheimer RD. Dermatomyositis: an overview of recent progress with emphasis on dermatologic aspects. Dermatol Clin. 2002;20(3):387–408.
Sontheimer RD. Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis sine myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J Am Acad Dermatol. 2002;46(4):626–36.
Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54(4):597–613. https://doi.org/10.1016/j.jaad.2005.10.041.
Femia AN, Vleugels RA, Callen JP. Cutaneous dermatomyositis: an updated review of treatment options and internal associations. Am J Clin Dermatol. 2013;14(4):291–313. https://doi.org/10.1007/s40257-013-0028-6.
Klein RQ, Teal V, Taylor L, Troxel AB, Werth VP. Number, characteristics, and classification of patients with dermatomyositis seen by dermatology and rheumatology departments at a large tertiary medical center. J Am Acad Dermatol. 2007;57(6):937–43. https://doi.org/10.1016/j.jaad.2007.08.024.
Leatham H, Schadt C, Chisolm S, Fretwell D, Chung L, Callen JP, et al. Evidence supports blind screening for internal malignancy in dermatomyositis. Medicine. 2018;97(2):e9639. https://doi.org/10.1097/md.0000000000009639.
Goreshi R, Chock M, Foering K, Feng R, Okawa J, Rose M, et al. Quality of life in dermatomyositis. J Am Acad Dermatol. 2011;65(6):1107–16. https://doi.org/10.1016/j.jaad.2010.10.016.
Wolstencroft PW, Chung L, Li S, Casciola-Rosen L, Fiorentino DF. Factors associated with clinical remission of skin disease in dermatomyositis. JAMA Dermatol. 2018;154(1):44–51. https://doi.org/10.1001/jamadermatol.2017.3758.
Da Silva DM, Patel B, Werth VP. Dermatomyositis: a diagnostic dilemma. J Am Acad Dermatol. 2018;79(2):371–3. https://doi.org/10.1016/j.jaad.2017.12.074.
Sigurgeirsson B, Lindelof B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis: a population-based study. N Engl J Med. 1992;326(6):363–7. https://doi.org/10.1056/NEJM199202063260602.
Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282–91. https://doi.org/10.3899/jrheum.140566.
Smith ES, Hallman JR, DeLuca AM, Goldenberg G, Jorizzo JL, Sangueza OP. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009;31(1):61–7. https://doi.org/10.1097/DAD.0b013e31818520e1.
Day J, Patel S, Limaye V. The role of magnetic resonance imaging techniques in evaluation and management of the idiopathic inflammatory myopathies. Semin Arthritis Rheum. 2017;46(5):642–9. https://doi.org/10.1016/j.semarthrit.2016.11.001.
Betteridge ZE, Gunawardena H, Chinoy H, North J, Ollier WE, Cooper RG, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis. 2009;68(10):1621–5. https://doi.org/10.1136/ard.2008.097162.
Fiorentino D, Casciola-Rosen L. Autoantibodies to transcription intermediary factor 1 in dermatomyositis shed insight into the cancer-myositis connection. Arthritis Rheum. 2012;64(2):346–9. https://doi.org/10.1002/art.33402.
Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65(1):25–34. https://doi.org/10.1016/j.jaad.2010.09.016.
Fujimoto M, Matsushita T, Hamaguchi Y, Kaji K, Asano Y, Ogawa F, et al. Autoantibodies to small ubiquitin-like modifier activating enzymes in Japanese patients with dermatomyositis: comparison with a UK Caucasian cohort. Ann Rheum Dis. 2013;72(1):151–3. https://doi.org/10.1136/annrheumdis-2012-201736.
Fujimoto M, Watanabe R, Ishitsuka Y, Okiyama N. Recent advances in dermatomyositis-specific autoantibodies. Curr Opin Rheumatol. 2016;28(6):636–44. https://doi.org/10.1097/BOR.0000000000000329.
Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78(4):776–85. https://doi.org/10.1016/j.jaad.2017.12.010.
Doyle TJ, Dellaripa PF. Lung manifestations in the rheumatic diseases. Chest. 2017;152(6):1283–95. https://doi.org/10.1016/j.chest.2017.05.015.
Mecoli CA, Christopher-Stine L. Management of interstitial lung disease in patients with myositis specific autoantibodies. Curr Rheumatol Rep. 2018;20(5):27. https://doi.org/10.1007/s11926-018-0731-7.
Morganroth PA, Kreider ME, Okawa J, Taylor L, Werth VP. Interstitial lung disease in classic and skin-predominant dermatomyositis: a retrospective study with screening recommendations. Arch Dermatol. 2010;146(7):729–38. https://doi.org/10.1001/archdermatol.2010.134.
Wolstencroft PW, Fiorentino DF. Dermatomyositis clinical and pathological phenotypes associated with myositis-specific autoantibodies. Curr Rheumatol Rep. 2018;20(5):28. https://doi.org/10.1007/s11926-018-0733-5.
Tartar DM, Chung L, Fiorentino DF. Clinical significance of autoantibodies in dermatomyositis and systemic sclerosis. Clin Dermatol. 2018;36(4):508–24. https://doi.org/10.1016/j.clindermatol.2018.04.008.
Hamaguchi Y, Kuwana M, Hoshino K, Hasegawa M, Kaji K, Matsushita T, et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol. 2011;147(4):391–8. https://doi.org/10.1001/archdermatol.2011.52.
Srivastava P, Dwivedi S, Misra R. Myositis-specific and myositis-associated autoantibodies in Indian patients with inflammatory myositis. Rheumatol Int. 2016;36(7):935–43. https://doi.org/10.1007/s00296-016-3494-3.
Komura K, Fujimoto M, Matsushita T, Kaji K, Kondo M, Hirano T, et al. Prevalence and clinical characteristics of anti-Mi-2 antibodies in Japanese patients with dermatomyositis. J Dermatol Sci. 2005;40(3):215–7. https://doi.org/10.1016/j.jdermsci.2005.09.004.
Petri MH, Satoh M, Martin-Marquez BT, Vargas-Ramirez R, Jara LJ, Saavedra MA, et al. Implications in the difference of anti-Mi-2 and -p155/140 autoantibody prevalence in two dermatomyositis cohorts from Mexico City and Guadalajara. Arthritis Res Ther. 2013;15(2):R48. https://doi.org/10.1186/ar4207.
Hengstman GJ, Vree Egberts WT, Seelig HP, Lundberg IE, Moutsopoulos HM, Doria A, et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Ann Rheum Dis. 2006;65(2):242–5. https://doi.org/10.1136/ard.2005.040717.
Aggarwal R, Loganathan P, Koontz D, Qi Z, Reed AM, Oddis CV. Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatology (Oxford). 2017;56(2):247–54. https://doi.org/10.1093/rheumatology/kew396.
Bodoki L, Nagy-Vincze M, Griger Z, Betteridge Z, Szollosi L, Danko K. Four dermatomyositis-specific autoantibodies-anti-TIF1gamma, anti-NXP2, anti-SAE and anti-MDA5-in adult and juvenile patients with idiopathic inflammatory myopathies in a Hungarian cohort. Autoimmun Rev. 2014;13(12):1211–9. https://doi.org/10.1016/j.autrev.2014.08.011.
Muro Y, Sugiura K, Akiyama M. Low prevalence of anti-small ubiquitin-like modifier activating enzyme antibodies in dermatomyositis patients. Autoimmunity. 2013;46(4):279–84. https://doi.org/10.3109/08916934.2012.755958.
Ge Y, Lu X, Shu X, et al. Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci Rep. 2017;7:188.
Tarricone E, Ghirardello A, Rampudda M, Bassi N, Punzi L, Doria A. Anti-SAE antibodies in autoimmune myositis: identification by unlabelled protein immunoprecipitation in an Italian patient cohort. J Immunol Methods. 2012;384(1–2):128–34. https://doi.org/10.1016/j.jim.2012.07.019.
Hamaguchi Y, Fujimoto M, Matsushita T, Kaji K, Komura K, Hasegawa M, et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PLoS One. 2013;8(4):e60442. https://doi.org/10.1371/journal.pone.0060442.
Lega JC, Fabien N, Reynaud Q, Durieu I, Durupt S, Dutertre M, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev. 2014;13(9):883–91. https://doi.org/10.1016/j.autrev.2014.03.004.
Hozumi H, Fujisawa T, Nakashima R, Johkoh T, Sumikawa H, Murakami A, et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir Med. 2016;121:91–9. https://doi.org/10.1016/j.rmed.2016.10.019.
Chen Z, Hu W, Wang Y, Guo Z, Sun L, Kuwana M. Distinct profiles of myositis-specific autoantibodies in Chinese and Japanese patients with polymyositis/dermatomyositis. Clin Rheumatol. 2015;34(9):1627–31. https://doi.org/10.1007/s10067-015-2935-9.
Li L, Wang Q, Wen X, Liu C, Wu C, Yang F, et al. Assessment of anti-MDA5 antibody as a diagnostic biomarker in patients with dermatomyositis-associated interstitial lung disease or rapidly progressive interstitial lung disease. Oncotarget. 2017;8(44):76129–40. https://doi.org/10.18632/oncotarget.19050.
Hall JC, Casciola-Rosen L, Samedy LA, Werner J, Owoyemi K, Danoff SK, et al. Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res (Hoboken). 2013;65(8):1307–15. https://doi.org/10.1002/acr.21992.
Fiorentino DF, Kuo K, Chung L, Zaba L, Li S, Casciola-Rosen L. Distinctive cutaneous and systemic features associated with antitranscriptional intermediary factor-1γ antibodies in adults with dermatomyositis. J Am Acad Dermatol. 2015;72(3):449–55. https://doi.org/10.1016/j.jaad.2014.12.009.
Fujimoto M, Hamaguchi Y, Kaji K, Matsushita T, Ichimura Y, Kodera M, et al. Myositis-specific anti-155/140 autoantibodies target transcription intermediary factor 1 family proteins. Arthritis Rheum. 2012;64(2):513–22. https://doi.org/10.1002/art.33403.
Meisterfeld S, Rober N, Conrad K, Beissert S, Aringer M, Gunther C. A chronic recurrent disease course of dermatomyositis is associated with autoantibodies against transcriptional intermediary factor 1-gamma. Br J Dermatol. 2017;177(2):590–3. https://doi.org/10.1111/bjd.15468.
Wolstencroft PW, Casciola-Rosen L, Fiorentino DF. Association between autoantibody phenotype and cutaneous adverse reactions to hydroxychloroquine in dermatomyositis. JAMA Dermatol. 2018;154(10):1199–203. https://doi.org/10.1001/jamadermatol.2018.2549.
Kaji K, Fujimoto M, Hasegawa M, Kondo M, Saito Y, Komura K, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford). 2007;46(1):25–8. https://doi.org/10.1093/rheumatology/kel161.
Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O’Hanlon TP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006;54(11):3682–9. https://doi.org/10.1002/art.22164.
Trallero-Araguas E, Rodrigo-Pendas JA, Selva-O’Callaghan A, Martinez-Gomez X, Bosch X, Labrador-Horrillo M, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis Rheum. 2012;64(2):523–32. https://doi.org/10.1002/art.33379.
Bernet LL, Lewis MA, Rieger KE, Casciola-Rosen L, Fiorentino DF. Ovoid palatal patch in dermatomyositis: a novel finding associated with anti-TIF1gamma (p155) antibodies. JAMA Dermatol. 2016;152(9):1049–51. https://doi.org/10.1001/jamadermatol.2016.1429.
Rogers A, Chung L, Li S, Casciola-Rosen L, Fiorentino DF. Cutaneous and systemic findings associated with nuclear matrix protein 2 antibodies in adult dermatomyositis patients. Arthritis Care Res (Hoboken). 2017;69(12):1909–14. https://doi.org/10.1002/acr.23210.
Albayda J, Pinal-Fernandez I, Huang W, Parks C, Paik J, Casciola-Rosen L, et al. Antinuclear matrix protein 2 autoantibodies and edema, muscle disease, and malignancy risk in dermatomyositis patients. Arthritis Care Res (Hoboken). 2017;69(11):1771–6. https://doi.org/10.1002/acr.23188.
Valenzuela A, Chung L, Casciola-Rosen L, Fiorentino D. Identification of clinical features and autoantibodies associated with calcinosis in dermatomyositis. JAMA Dermatol. 2014;150(7):724–9. https://doi.org/10.1001/jamadermatol.2013.10416.
Ishikawa A, Muro Y, Sugiura K, Akiyama M. Development of an ELISA for detection of autoantibodies to nuclear matrix protein 2. Rheumatology (Oxford). 2012;51(7):1181–7. https://doi.org/10.1093/rheumatology/kes033.
Buchbinder R, Forbes A, Hall S, Dennett X, Giles G. Incidence of malignant disease in biopsy-proven inflammatory myopathy: a population-based cohort study. Ann Intern Med. 2001;134(12):1087–95.
Chow WH, Gridley G, Mellemkjaer L, McLaughlin JK, Olsen JH, Fraumeni JF Jr. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark. Cancer Causes Control. 1995;6(1):9–13.
Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96–100. https://doi.org/10.1016/S0140-6736(00)03540-6.
Stockton D, Doherty VR, Brewster DH. Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a Scottish population-based cohort study. Br J Cancer. 2001;85(1):41–5. https://doi.org/10.1054/bjoc.2001.1699.
Chen YJ, Wu CY, Huang YL, Wang CB, Shen JL, Chang YT. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. 2010;12(2):R70. https://doi.org/10.1186/ar2987.
Lu X, Yang H, Shu X, Chen F, Zhang Y, Zhang S, et al. Factors predicting malignancy in patients with polymyositis and dermatomyostis: a systematic review and meta-analysis. PLoS One. 2014;9(4):e94128. https://doi.org/10.1371/journal.pone.0094128.
Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li S, Mammen AL, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum. 2013;65(11):2954–62. https://doi.org/10.1002/art.38093.
Quain RD, Werth VP. Management of cutaneous dermatomyositis: current therapeutic options. Am J Clin Dermatol. 2006;7(6):341–51. https://doi.org/10.2165/00128071-200607060-00002.
Hundley JL, Carroll CL, Lang W, Snively B, Yosipovitch G, Feldman SR, et al. Cutaneous symptoms of dermatomyositis significantly impact patients’ quality of life. J Am Acad Dermatol. 2006;54(2):217–20. https://doi.org/10.1016/j.jaad.2004.12.015.
Anyanwu CO, Fiorentino DF, Chung L, Dzuong C, Wang Y, Okawa J, et al. Validation of the Cutaneous Dermatomyositis Disease Area and Severity Index: characterizing disease severity and assessing responsiveness to clinical change. Br J Dermatol. 2015;173(4):969–74. https://doi.org/10.1111/bjd.13915.
Chansky PB, Olazagasti JM, Feng R, Werth VP. Cutaneous dermatomyositis disease course followed over time using the Cutaneous Dermatomyositis Disease Area and Severity Index. J Am Acad Dermatol. 2018;79(3):464–9. https://doi.org/10.1016/j.jaad.2017.10.022(e2).
Yassaee M, Fiorentino D, Okawa J, Taylor L, Coley C, Troxel AB, et al. Modification of the cutaneous dermatomyositis disease area and severity index, an outcome instrument. Br J Dermatol. 2010;162(3):669–73. https://doi.org/10.1111/j.1365-2133.2009.09521.x.
Cheong WK, Hughes GR, Norris PG, Hawk JL. Cutaneous photosensitivity in dermatomyositis. Br J Dermatol. 1994;131(2):205–8.
Lam C, Vleugels RA. Management of cutaneous dermatomyositis. Dermatol Ther. 2012;25(2):112–34. https://doi.org/10.1111/j.1529-8019.2012.01491.x.
Callen JP, Wortmann RL. Dermatomyositis. Clin Dermatol. 2006;24(5):363–73. https://doi.org/10.1016/j.clindermatol.2006.07.001.
Shirani Z, Kucenic MJ, Carroll CL, Fleischer AB Jr, Feldman SR, Yosipovitch G, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29(3):273–6. https://doi.org/10.1111/j.1365-2230.2004.01510.x.
Kim HJ, Zeidi M, Bonciani D, Pena SM, Tiao J, Sahu S, et al. Itch in dermatomyositis: the role of increased skin interleukin-31. Br J Dermatol. 2018;179(3):669–78. https://doi.org/10.1111/bjd.16498.
Werth VP, Hejazi E, Pena SM, Haber JS, Okawa J, Feng R, et al. FRI0470—a phase 2 study of safety and efficacy of lenabasum (JBT-101), a cannabinoid receptor type 2 agonist, in refractory skin-predominant dermatomyositis. Ann Rheum Dis. 2018;77(Suppl. 2):763–4. https://doi.org/10.1136/annrheumdis-2018-eular.3531.
Werth VP, Patel B, Concha JS, Okawa J, Pearson D, Hejazi E, et al. SAT0512—safety and efficacy of lenabasum in refractory skin-predominant dermatomyositis subjects treated in an open label extension of trial jbt101-dm-001. Ann Rheum Dis. 2018;77(Suppl. 2):1111–2. https://doi.org/10.1136/annrheumdis-2018-eular.5629.
Yoshimasu T, Ohtani T, Sakamoto T, Oshima A, Furukawa F. Topical FK506 (tacrolimus) therapy for facial erythematous lesions of cutaneous lupus erythematosus and dermatomyositis. Eur J Dermatol. 2002;12(1):50–2.
Ueda M, Makinodan R, Matsumura M, Ichihashi M. Successful treatment of amyopathic dermatomyositis with topical tacrolimus. Br J Dermatol. 2003;148(3):595–6.
Lampropoulos CE, D’Cruz DP. Topical tacrolimus treatment in a patient with dermatomyositis. Ann Rheum Dis. 2005;64(9):1376–7. https://doi.org/10.1136/ard.2004.032714.
Hollar CB, Jorizzo JL. Topical tacrolimus 0.1% ointment for refractory skin disease in dermatomyositis: a pilot study. J Dermatol Treat. 2004;15(1):35–9. https://doi.org/10.1080/09541440042000269.
Kim JE, Jeong MG, Lee HE, Ko JY, Ro YS. Successful treatment of cutaneous lesions of dermatomyositis with topical pimecrolimus. Ann Dermatol. 2011;23(3):348–51. https://doi.org/10.5021/ad.2011.23.3.348.
Garcia-Doval I, Cruces M. Topical tacrolimus in cutaneous lesions of dermatomyositis: lack of effect in side-by-side comparison in five patients. Dermatology. 2004;209(3):247–8. https://doi.org/10.1159/000079903.
Ring J, Mohrenschlager M, Henkel V. The US FDA ‘black box’ warning for topical calcineurin inhibitors: an ongoing controversy. Drug Saf. 2008;31(3):185–98. https://doi.org/10.2165/00002018-200831030-00001.
Soter NA, Fleischer AB Jr, Webster GF, Monroe E, Lawrence I. Tacrolimus ointment for the treatment of atopic dermatitis in adult patients: part II, safety. J Am Acad Dermatol. 2001;44(1 Suppl.):S39–46.
Ochsendorf FR. Use of antimalarials in dermatology. Journal der Deutschen Dermatologischen Gesellschaft. 2010;8(10):829–44. https://doi.org/10.1111/j.1610-0387.2010.07490.x(quiz 45).
Woo TY, Callen JP, Voorhees JJ, Bickers DR, Hanno R, Hawkins C. Cutaneous lesions of dermatomyositis are improved by hydroxychloroquine. J Am Acad Dermatol. 1984;10(4):592–600.
James WD, Dawson N, Rodman OG. The treatment of dermatomyositis with hydroxchloroquine. J Rheumatol. 1985;12(6):1214–6.
Cosnes A, Amaudric F, Gherardi R, Verroust J, Wechsler J, Revuz J, et al. Dermatomyositis without muscle weakness: long-term follow-up of 12 patients without systemic corticosteroids. Arch Dermatol. 1995;131(12):1381–5.
Cox NH. Amyopathic dermatomyositis, photosensitivity and hydroxychloroquine. Br J Dermatol. 1995;132(6):1016–7.
Dawkins MA, Jorizzo JL, Walker FO, Albertson D, Sinal SH, Hinds A. Dermatomyositis: a dermatology-based case series. J Am Acad Dermatol. 1998;38(3):397–404.
Pinard J, Femia AN, Roman M, Alsarheed A, Joyce C, Lin J, et al. Systemic treatment for clinically amyopathic dermatomyositis at 4 tertiary care centers. JAMA Dermatol. 2019;155(4):494–6. https://doi.org/10.1001/jamadermatol.2018.5215.
Pelle MT, Callen JP. Adverse cutaneous reactions to hydroxychloroquine are more common in patients with dermatomyositis than in patients with cutaneous lupus erythematosus. Arch Dermatol. 2002;138(9):1231–3 (discussion 3).
Anyanwu CO, Chansky PB, Feng R, Carr K, Okawa J, Werth VP. The systemic management of cutaneous dermatomyositis: results of a stepwise strategy. Int J Womens Dermatol. 2017;3(4):189–94. https://doi.org/10.1016/j.ijwd.2017.05.001.
Ang GC, Werth VP. Combination antimalarials in the treatment of cutaneous dermatomyositis: a retrospective study. Arch Dermatol. 2005;141(7):855–9. https://doi.org/10.1001/archderm.141.7.855.
Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF, American Academy of Ophthalmology. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016;123(6):1386–94. https://doi.org/10.1016/j.ophtha.2016.01.058.
Estes ML, Ewing-Wilson D, Chou SM, Mitsumoto H, Hanson M, Shirey E, et al. Chloroquine neuromyotoxicity: clinical and pathologic perspective. Am J Med. 1987;82(3):447–55. https://doi.org/10.1016/0002-9343(87)90444-x.
Cronstein BN, Naime D, Ostad E. The antiinflammatory effects of methotrexate are mediated by adenosine. Adv Exp Med Biol. 1994;370:411–6.
Dalakas MC. Inflammatory myopathies: management of steroid resistance. Curr Opin Neurol. 2011;24(5):457–62. https://doi.org/10.1097/WCO.0b013e32834a9589.
Malaviya AN, Many A, Schwartz RS. Treatment of dermatomyositis with methotrexate. Lancet. 1968;2(7566):485–8.
Newman ED, Scott DW. The use of low-dose oral methotrexate in the treatment of polymyositis and dermatomyositis. J Clin Rheumatol. 1995;1(2):99–102.
Zieglschmid-Adams ME, Pandya AG, Cohen SB, Sontheimer RD. Treatment of dermatomyositis with methotrexate. J Am Acad Dermatol. 1995;32(5 Pt 1):754–7.
Kasteler JS, Callen JP. Low-dose methotrexate administered weekly is an effective corticosteroid-sparing agent for the treatment of the cutaneous manifestations of dermatomyositis. J Am Acad Dermatol. 1997;36(1):67–71.
Zieglschmid-Adams ME, Pandya A, Cohen SB, Sontheimer RD. The value of methotrexate in dermatomyositis. J Am Acad Dermatol. 1998;38(1):130–2.
Hornung T, Ko A, Tuting T, Bieber T, Wenzel J. Efficacy of low-dose methotrexate in the treatment of dermatomyositis skin lesions. Clin Exp Dermatol. 2012;37(2):139–42. https://doi.org/10.1111/j.1365-2230.2011.04188.x.
Cannon GW. Methotrexate pulmonary toxicity. Rheum Dis Clin N Am. 1997;23(4):917–37.
Hoekstra M, Haagsma C, Neef C, Proost J, Knuif A, van de Laar M. Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J Rheumatol. 2004;31(4):645–8.
Dalakas MC. Immunotherapy of inflammatory myopathies: practical approach and future prospects. Curr Treat Options Neurol. 2011;13(3):311–23. https://doi.org/10.1007/s11940-011-0119-8.
Drake LA, Dinehart SM, Farmer ER, Goltz RW, Graham GF, Hordinsky MK, et al. Guidelines of care for dermatomyositis. American Academy of Dermatology. J Am Acad Dermatol. 1996;34(5 Pt 1):824–9.
Vleugels RA, Callen JP. Dermatomyositis: current and future treatments. Expert Rev Dermatol. 2009;4:581–94.
Orvis AK, Wesson SK, Breza TS Jr, Church AA, Mitchell CL, Watkins SW. Mycophenolate mofetil in dermatology. J Am Acad Dermatol. 2009;60(2):183–99. https://doi.org/10.1016/j.jaad.2008.08.049(quiz 200–2).
Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47(2–3):85–118.
Gelber AC, Nousari HC, Wigley FM. Mycophenolate mofetil in the treatment of severe skin manifestations of dermatomyositis: a series of 4 cases. J Rheumatol. 2000;27(6):1542–5.
Majithia V, Harisdangkul V. Mycophenolate mofetil (CellCept): an alternative therapy for autoimmune inflammatory myopathy. Rheumatology (Oxford). 2005;44(3):386–9. https://doi.org/10.1093/rheumatology/keh499.
Edge JC, Outland JD, Dempsey JR, Callen JP. Mycophenolate mofetil as an effective corticosteroid-sparing therapy for recalcitrant dermatomyositis. Arch Dermatol. 2006;142(1):65–9. https://doi.org/10.1001/archderm.142.1.65.
Rowin J, Amato AA, Deisher N, Cursio J, Meriggioli MN. Mycophenolate mofetil in dermatomyositis: is it safe? Neurology. 2006;66(8):1245–7. https://doi.org/10.1212/01.wnl.0000208416.32471.c0.
Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken). 2010;62(10):1496–501. https://doi.org/10.1002/acr.20212.
Tsuchiya H, Tsuno H, Inoue M, Takahashi Y, Yamashita H, Kaneko H, et al. Mycophenolate mofetil therapy for rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis. Mod Rheumatol. 2014;24(4):694–6. https://doi.org/10.3109/14397595.2013.874762.
Hisanaga J, Kotani T, Fujiki Y, Yoshida S, Takeuchi T, Makino S. Successful multi-target therapy including rituximab and mycophenolate mofetil in anti-melanoma differentiation-associated gene 5 antibody-positive rapidly progressive interstitial lung disease with clinically amyopathic dermatomyositis. Int J Rheum Dis. 2017;20(12):2182–5. https://doi.org/10.1111/1756-185X.13136.
Koyama RVL, Braga TKK, da Silva Dias GA, Fujihara S, Fuzii HT, Yoshikawa GT. Hypomyopathic dermatomyositis associated with interstitial lung disease and good response to mycophenolate mofetil: case-based review. Clin Rheumatol. 2017;36(8):1919–26. https://doi.org/10.1007/s10067-017-3671-0.
Morisset J, Johnson C, Rich E, Collard HR, Lee JS. Management of myositis-related interstitial lung disease. Chest. 2016;150(5):1118–28. https://doi.org/10.1016/j.chest.2016.04.007.
Dalakas MC. Mechanistic effects of IVIg in neuroinflammatory diseases: conclusions based on clinicopathologic correlations. J Clin Immunol. 2014;34(Suppl. 1):S120–6. https://doi.org/10.1007/s10875-014-0024-5.
Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329(27):1993–2000. https://doi.org/10.1056/NEJM199312303292704.
Femia AN, Eastham AB, Lam C, Merola JF, Qureshi AA, Vleugels RA. Intravenous immunoglobulin for refractory cutaneous dermatomyositis: a retrospective analysis from an academic medical center. J Am Acad Dermatol. 2013;69(4):654–7. https://doi.org/10.1016/j.jaad.2013.06.007.
Bounfour T, Bouaziz JD, Bezier M, Cordoliani F, Saussine A, Petit A, et al. Clinical efficacy of intravenous immunoglobulins for the treatment of dermatomyositis skin lesions without muscle disease. J Eur Acad Dermatol Venereol. 2014;28(9):1150–7. https://doi.org/10.1111/jdv.12223.
Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65(2):314–24. https://doi.org/10.1002/art.37754.
Tokunaga K, Hagino N. Dermatomyositis with rapidly progressive interstitial lung disease treated with rituximab: a report of 3 cases in Japan. Intern Med. 2017;56(11):1399–403. https://doi.org/10.2169/internalmedicine.56.7956.
Ogawa Y, Kishida D, Shimojima Y, Hayashi K, Sekijima Y. Effective administration of rituximab in anti-MDA5 antibody-positive dermatomyositis with rapidly progressive interstitial lung disease and refractory cutaneous involvement: a case report and literature review. Case Rep Rheumatol. 2017;2017:5386797. https://doi.org/10.1155/2017/5386797.
Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52(2):601–7. https://doi.org/10.1002/art.20849.
Chung L, Genovese MC, Fiorentino DF. A pilot trial of rituximab in the treatment of patients with dermatomyositis. Arch Dermatol. 2007;143(6):763–7. https://doi.org/10.1001/archderm.143.6.763.
Shreberk-Hassidim R, Ramot Y, Zlotogorski A. Janus kinase inhibitors in dermatology: a systematic review. J Am Acad Dermatol. 2017;76(4):745–53. https://doi.org/10.1016/j.jaad.2016.12.004(e19).
Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76(4):736–44. https://doi.org/10.1016/j.jaad.2016.12.005.
Hornung T, Janzen V, Heidgen FJ, Wolf D, Bieber T, Wenzel J. Remission of recalcitrant dermatomyositis treated with ruxolitinib. N Engl J Med. 2014;371(26):2537–8. https://doi.org/10.1056/NEJMc1412997.
Kurtzman DJ, Wright NA, Lin J, Femia AN, Merola JF, Patel M, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: an alternative treatment. JAMA Dermatol. 2016;152(8):944–5. https://doi.org/10.1001/jamadermatol.2016.0866.
Safety study of tofacitinib versus tumor necrosis factor (TNF) inhibitor in subjects with rheumatoid arthritis. https://ClinicalTrials.gov/show/NCT02092467. Accessed 13 Jan 2020.
Kaposztas Z, Etheridge WB, Kahan BD. Case report: successful treatment of posttransplant lymphoproliferative disorder and quiescence of dermatomyositis with rituximab and sirolimus. Transplant Proc. 2008;40(5):1744–6. https://doi.org/10.1016/j.transproceed.2007.11.072.
Kim HJ, Hong YK, Yoo WH. Dermatomyositis, complicated with pneumomediastinum, successfully treated with cyclosporine A: a case report and review of literature. Rheumatol Int. 2009;29(9):1101–4. https://doi.org/10.1007/s00296-008-0822-2.
Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H, Devulder B, et al. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol. 2001;28(10):2230–7.
Meyer O, Hayem G, Palazzo E, Crestani B, Debray MP, Ballard M. Interstitial lung disease due to polymyositis or dermatomyositis: effect of a 6-month course of i.v. pulse cyclophosphamide. Clin Exp Rheumatol. 2005;23(5):724.
Mitsui T, Kuroda Y, Kunishige M, Matsumoto T. Successful treatment with tacrolimus in a case of refractory dermatomyositis. Intern Med. 2005;44(11):1197–9.
Nadiminti U, Arbiser JL. Rapamycin (sirolimus) as a steroid-sparing agent in dermatomyositis. J Am Acad Dermatol. 2005;52(2 Suppl. 1):17–9. https://doi.org/10.1016/j.jaad.2004.05.044.
Ochi S, Nanki T, Takada K, Suzuki F, Komano Y, Kubota T, et al. Favorable outcomes with tacrolimus in two patients with refractory interstitial lung disease associated with polymyositis/dermatomyositis. Clin Exp Rheumatol. 2005;23(5):707–10.
Ramirez G, Asherson RA, Khamashta MA, Cervera R, D’Cruz D, Hughes GR. Adult-onset polymyositis-dermatomyositis: description of 25 patients with emphasis on treatment. Semin Arthritis Rheum. 1990;20(2):114–20.
Sinoway PA, Callen JP. Chlorambucil: an effective corticosteroid-sparing agent for patients with recalcitrant dermatomyositis. Arthritis Rheum. 1993;36(3):319–24.
Vencovsky J, Jarosova K, Machacek S, Studynkova J, Kafkova J, Bartunkova J, et al. Cyclosporine A versus methotrexate in the treatment of polymyositis and dermatomyositis. Scand J Rheumatol. 2000;29(2):95–102.
Yamasaki Y, Yamada H, Yamasaki M, Ohkubo M, Azuma K, Matsuoka S, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford). 2007;46(1):124–30. https://doi.org/10.1093/rheumatology/kel112.
Shimojima Y, Ishii W, Kato T, Hoshi K, Matsuda M, Hashimoto T, et al. Intractable skin necrosis and interstitial pneumonia in amyopathic dermatomyositis, successfully treated with cyclosporin A. Intern Med. 2003;42(12):1253–8. https://doi.org/10.2169/internalmedicine.42.1253.
Yamada A, Ohshima Y, Omata N, Yasutomi M, Mayumi M. Steroid-sparing effect of tacrolimus in a patient with juvenile dermatomyositis presenting poor bioavailability of cyclosporine A. Eur J Pediatr. 2004;163(9):561–2. https://doi.org/10.1007/s00431-004-1497-7.
Martin Nalda A, Modesto Caballero C, Arnal Guimeral C, Boronat Rom M, Barcelo Garcia P. Efficacy of tacrolimus (FK-506) in the treatment of recalcitrant juvenile dermatomyositis: study of 6 cases. Med Clin (Barc). 2006;127(18):697–701.
Hassan J, van der Net JJ, van Royen-Kerkhof A. Treatment of refractory juvenile dermatomyositis with tacrolimus. Clin Rheumatol. 2008;27(11):1469–71. https://doi.org/10.1007/s10067-008-0973-2.
Tsujimura S, Saito K, Tanaka Y. Complete resolution of dermatomyositis with refractory cutaneous vasculitis by intravenous cyclophosphamide pulse therapy. Intern Med. 2008;47(21):1935–40.
Boswell JS, Costner MI. Leflunomide as adjuvant treatment of dermatomyositis. J Am Acad Dermatol. 2008;58(3):403–6. https://doi.org/10.1016/j.jaad.2007.08.014.
Callander J, Robson Y, Ingram J, Piguet V. Treatment of clinically amyopathic dermatomyositis in adults: a systematic review. Br J Dermatol. 2018;179(6):1248–55. https://doi.org/10.1111/bjd.14726.
Cohen JB. Cutaneous involvement of dermatomyositis can respond to dapsone therapy. Int J Dermatol. 2002;41(3):182–4.
Konohana A, Kawashima J. Successful treatment of dermatomyositis with dapsone. Clin Exp Dermatol. 1994;19(4):367.
Sangle VS, Sangle SR, D’Cruz DP. Leflunomide as a remission-maintaining therapy in difficult-to-treat dermatomyositis. Ann Rheum Dis. 2008;67(5):723. https://doi.org/10.1136/ard.2007.073221.
Sebastiani M, Puccini R, Manfredi A, Manni E, Colaci M, Mattei P, et al. Staphylococcus protein A-based extracorporeal immunoadsorption and thalidomide in the treatment of skin manifestation of dermatomyositis: a case report. Ther Apher Dial. 2009;13(3):225–8. https://doi.org/10.1111/j.1744-9987.2009.00689.x.
Zuber M, John S, Pfreundschuh M, Gause A. A young woman with a photosensitive pruritic rash on her face and upper trunk. Arthritis Rheum. 1996;39(8):1419–22.
Nahmias Z, Nambudiri VE, Vleugels RA. Thalidomide and lenalidomide for the treatment of refractory dermatologic conditions. J Am Acad Dermatol. 2016;75(1):210–2. https://doi.org/10.1016/j.jaad.2016.03.022.
Stirling DI. Thalidomide and its impact in dermatology. Semin Cutan Med Surg. 1998;17(4):231–42.
Norman R, Greenberg RG, Jackson JM. Case reports of etanercept in inflammatory dermatoses. J Am Acad Dermatol. 2006;54(3 Suppl. 2):S139–42. https://doi.org/10.1016/j.jaad.2005.11.1090.
Chen D, Wang XB, Zhou Y, Zhu XC. Efficacy of infliximab in the treatment for dermatomyositis with acute interstitial pneumonia: a study of fourteen cases and literature review. Rheumatol Int. 2013;33(10):2455–8. https://doi.org/10.1007/s00296-012-2653-4.
Dold S, Justiniano ME, Marquez J, Espinoza LR. Treatment of early and refractory dermatomyositis with infliximab: a report of two cases. Clin Rheumatol. 2007;26(7):1186–8. https://doi.org/10.1007/s10067-006-0325-z.
Rouster-Stevens KA, Ferguson L, Morgan G, Huang CC, Pachman LM. Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2014;66(5):783–7. https://doi.org/10.1002/acr.22198.
Muscle Study G. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol. 2011;70(3):427–36. https://doi.org/10.1002/ana.22477.
Brunasso AM, Scocco GL, Massone C. Dermatomyositis during adalimumab therapy for rheumatoid arthritis. J Rheumatol. 2010;37(7):1549–50. https://doi.org/10.3899/jrheum.091413.
Dastmalchi M, Grundtman C, Alexanderson H, Mavragani CP, Einarsdottir H, Helmers SB, et al. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann Rheum Dis. 2008;67(12):1670–7. https://doi.org/10.1136/ard.2007.077974.
Flendrie M, Vissers WH, Creemers MC, de Jong EM, van de Kerkhof PC, van Riel PL. Dermatological conditions during TNF-alpha-blocking therapy in patients with rheumatoid arthritis: a prospective study. Arthritis Res Ther. 2005;7(3):R666–76. https://doi.org/10.1186/ar1724.
Hall HA, Zimmermann B. Evolution of dermatomyositis during therapy with a tumor necrosis factor alpha inhibitor. Arthritis Rheum. 2006;55(6):982–4. https://doi.org/10.1002/art.22358.
Hengstman GJ, De Bleecker JL, Feist E, Vissing J, Denton CP, Manoussakis MN, et al. Open-label trial of anti-TNF-alpha in dermato- and polymyositis treated concomitantly with methotrexate. Eur Neurol. 2008;59(3–4):159–63. https://doi.org/10.1159/000114036.
Iannone F, Scioscia C, Falappone PC, Covelli M, Lapadula G. Use of etanercept in the treatment of dermatomyositis: a case series. J Rheumatol. 2006;33(9):1802–4.
Ishiguro T, Takayanagi N, Miyahara Y, Yanagisawa T, Sugita Y. Antisynthetase (anti PL-7 antibody) syndrome presenting as a skin rash and exacerbation of interstitial pneumonia during treatment for rheumatoid arthritis. Nihon Kokyuki Gakkai Zasshi. 2010;48(3):240–6.
Ishikawa Y, Yukawa N, Kawabata D, Ohmura K, Fujii T, Usui T, et al. A case of antisynthetase syndrome in a rheumatoid arthritis patient with anti-PL-12 antibody following treatment with etanercept. Clin Rheumatol. 2011;30(3):429–32. https://doi.org/10.1007/s10067-010-1666-1.
Ishikawa Y, Yukawa N, Ohmura K, Hosono Y, Imura Y, Kawabata D, et al. Etanercept-induced anti-Jo-1-antibody-positive polymyositis in a patient with rheumatoid arthritis: a case report and review of the literature. Clin Rheumatol. 2010;29(5):563–6. https://doi.org/10.1007/s10067-009-1370-1.
Klein R, Rosenbach M, Kim EJ, Kim B, Werth VP, Dunham J. Tumor necrosis factor inhibitor-associated dermatomyositis. Arch Dermatol. 2010;146(7):780–4. https://doi.org/10.1001/archdermatol.2010.142.
Liozon E, Ouattara B, Loustaud-Ratti V, Vidal E. Severe polymyositis and flare in autoimmunity following treatment with adalimumab in a patient with overlapping features of polyarthritis and scleroderma. Scand J Rheumatol. 2007;36(6):484–6. https://doi.org/10.1080/03009740701281293.
Liu SW, Velez NF, Lam C, Femia A, Granter SR, Townsend HB, et al. Dermatomyositis induced by anti-tumor necrosis factor in a patient with juvenile idiopathic arthritis. JAMA Dermatol. 2013;149(10):1204–8. https://doi.org/10.1001/jamadermatol.2013.5220.
Musial J, Undas A, Celinska-Lowenhoff M. Polymyositis associated with infliximab treatment for rheumatoid arthritis. Rheumatology (Oxford). 2003;42(12):1566–8. https://doi.org/10.1093/rheumatology/keg388.
Riolo G, Towheed TE. Anti-tumor necrosis factor inhibitor therapy-induced dermatomyositis and fasciitis. J Rheumatol. 2012;39(1):192–4.
Urata Y, Wakai Y, Kowatari K, Nitobe T, Mizushima Y. Polymyositis associated with infliximab treatment for rheumatoid arthritis. Mod Rheumatol. 2006;16(6):410–1. https://doi.org/10.1007/s10165-006-0523-1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this review.
Conflict of interest
Gabriela A. Cobos has no conflicts of interest that are directly relevant to the content of this article. Alisa Femia is a principal investigator for a Pfizer-sponsored trial on dermatomyositis treatment. Ruth Ann Vleugels is a principal investigator for a Pfizer-sponsored trial on dermatomyositis treatment.
Rights and permissions
About this article

Cite this article
Cobos, G.A., Femia, A. & Vleugels, R.A. Dermatomyositis: An Update on Diagnosis and Treatment. Am J Clin Dermatol 21, 339–353 (2020). https://doi.org/10.1007/s40257-020-00502-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-020-00502-6