
Abstract
Abrupt discontinuation of many drugs used in medicine causes withdrawal syndromes, some of which can be fatal. Discontinuation of a number of cardiovascular drugs can increase the risk of cardiovascular events. Whereas aspirin administration is known to decrease the risk of vascular ischemic problems, aspirin withdrawal may temporarily increase the risk of thrombotic events. Indeed, aspirin withdrawal has been associated with an increased risk of thrombosis both in clinical and fundamental research studies. Such complications occur within the first month after interrupting aspirin therapy and their mechanism remains unexplained. We have previously demonstrated that aspirin, when injected as a single high dose (100 mg/kg), induces a prothrombotic state in the rat, similar to that described above, 8 and 10 days after administration. This effect in the rat may be reproduced 1 hour after a single injection of ultra-low-dose aspirin. Caution is therefore required regarding the possibility of drug discontinuation effects within the framework of drug safety evaluation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Aspirin remains the most widely used drug for the prevention of vascular events. It has shown a positive effect in patients with acute myocardial infarction and in the prevention of atherothrombotic events [1, 2]. Recent observational epidemiological evidence has raised the concern that aspirin withdrawal can carry an increased thrombotic risk. This risk appears to be increased for ischemic stroke [3–5], cardiovascular problems [6–12], and acute lower limb ischemia [13]. The delay between aspirin discontinuation and the thrombotic event was between 7 and 30 days in most reports and most commonly between 7 and 10 days. The stroke occurred 7.4 ± 1.26 days after discontinuation of the treatment. Fischer et al. [6] reported a 1.52-fold risk of acute myocardial infarction (95 % confidence interval 1.33–1.74) in patients who stopped taking non-steroidal anti-inflammatory drugs (NSAIDs) between 1 and 29 days before testing compared with non-users. Ferrari et al. [7] supports the hypothesis that aspirin withdrawal may represent a real risk for the occurrence of a new coronary event. Eisenberg et al. [14] showed that the discontinuation of aspirin increased the risk of drug-eluting stent thrombosis around day 8 after discontinuation.
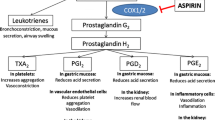
Cyclooxygenase-1 (COX-1) catalyzes the transformation of arachidonic acid to the unstable intermediate prostaglandin PGH2. Subsequently, thromboxane synthase acts on PGH2 to form TXA2, a transient biological product that induces platelet aggregation and has powerful vasoconstrictor effects. Aspirin acts primarily by interfering with the biosynthesis of the cyclic prostanoids TXA2, prostacyclin, and other prostaglandins. It irreversibly inhibits COX-1 by acetylation of serine-530 and induces a long-lasting functional defect in platelet function. The resultant decrease in production of prostaglandins and TXA2 probably accounts for much of aspirin’s antithrombotic effect [15, 16]. The plasma half-life of aspirin is only 20 min in circulating blood. Thereafter, it is rapidly deacetylated and converted to salicylate in vivo. Salicylate does not affect COX-1 or COX-2 activity [17].
Because platelets cannot generate new COX, the effects of aspirin last for the duration of the life of the platelet (8 days). After a single dose of aspirin, platelet COX activity recovers by 10 % per day in parallel with platelet turnover. Although it may take 8 days for the total platelet population to be renewed, it has been shown that if as few as 20 % of the platelets have normal COX activity, hemostasis may be normal.
TXA2 and PGI2 have opposing effects on hemostasis, but the data suggest that the antithrombotic effects of TXA2 inhibition predominate over the possible prothrombotic effects of PGI2 inhibition [15].
The usual antithrombotic dose of acetylsalicylic acid (ASA) ranges from 80 to 500 mg/day. Platelet inhibition, as indicated by aggregometry, occurs very rapidly: within 5 min of ingestion of lysine acetylsalicylate 320 mg and within 30–60 min of oral administration of ASA 320 mg [18].
As COX-1 inhibition by aspirin is irreversible, there is a cumulative inhibition of TXA2 generation by platelets when low doses of aspirin are administered over a long time [19]. There is a non-linear relationship between inhibition of platelet TXA2 generation and inhibition of TXA2-dependent platelet aggregation. More than 95 % inhibition of TXA2 generation is necessary to influence platelet function [20]. With low-dose aspirin, platelet thromboxane falls below 95 % after only a few days. Nevertheless, both low-dose (40–60 mg) and high-dose (500 mg) aspirin suppresses thromboxane A2 synthesis by >95 % [21, 22].
Recently published surveys have warned about the increased risk of thrombosis after aspirin discontinuation, which includes acute coronary syndromes, stent-associated thrombosis, acute myocardial infarction, ischemic stroke, and lower limb ischemia [4–12].
The delay of several days observed between aspirin withdrawal and the thrombotic event raises the possibility that aspirin might have some long-lasting prothrombotic effect. The hypothesis of a reverse effect and different aspects of the prothrombotic effects of aspirin have been studied in our laboratory.
To observe in vivo the intimate nature of changes in the pathophysiology of thrombosis, endothelial cells, sub-endothelial collagen and platelets should be present in the most unmodified manner.
2 Experimental Investigations
2.1 Laser-Induced Thrombosis Model (LITM)
In 1993, we used a laser-induced thrombosis model (LITM) in mesenteric vessels [23]. This model allows the induction of localized endothelial destruction of single cells, leading to physiological blood disturbances, platelet activation and adhesion to the exposed sub-endothelium.
Vascular lesions were produced by argon laser. The laser beam was coaxially inserted into the induced light beam of a microscope. The wavelength used was 514.5 nm and the energy was adjusted to 120 mW. The exposure time for the laser shot was 1/15 s. Microscopic images were recorded by a videotape recorder through a video camera and monitored on the television screen. The dynamic course of thrombus formation was continuously monitored and the time of the experiment was recorded automatically.
The spot for a single laser shot was positioned on the endothelium at the border between the luminal portion of the vessel wall and the lumen of the arteriole. The results were evaluated by direct observation through the microscope and by recording the total sequence of intravascular reaction. At least three parameters were measured: (1) the number of laser injuries (LI) required to induce platelet thrombus formation (when no visible thrombus occurred after the first shot, a second, third, and fourth laser shot were applied to the vessel at the same site as the first one, 1, 5, and 8 min, respectively); (2) the number of emboli (NE) removed by the blood stream after thrombus formation; and (3) the duration of embolization (DE) in minutes.
To validate this model, higher doses of aspirin were initially tested (50, 100, and 200 mg/kg rat weight) and also ultra-low-dose aspirin (ULDA). The three high doses increased the number of LI required to start embolization in a dose-dependent manner, and the NE and the DE were significantly decreased [23, 24].
2.1.1 The Effect of Different Doses of Aspirin, Including Very Low Doses in an LITM
ASA dilution was prepared as follows: 1 g of pure, finely powdered ASA was suspended in 99 mL alcohol (70°). After vigorous shaking, 1 mL of this dilution was then mixed in 99 mL of distilled water and vigorously shaken. This process was repeated to the last dilution (see Table 1 for doses and groups distribution).
The products studied were saline control, salicylate 100 mg/kg, aspirin 100 mg/kg, and decreasing doses of aspirin covering a highly extended dose range. Aspirin 100 mg/kg bodyweight showed an antithrombotic effect with a decreased formation of emboli and DE. Importantly, the ULDA (10−30 and 10−60 mg/kg rat bodyweight) showed a prothrombotic effect increasing the NE and the DE (see Table 2) [25].
2.1.2 Study of Thromboembolic Complications Several Days After a Single-Dose Administration of Aspirin with LITM in Rats
This effect was studied with the injection of one antithrombotic dose of aspirin (100 mg/kg bodyweight) and by performing laser-induced thrombosis 2, 4, 6, 8, 10, 12, 14, and 16 days after the injection [26].
The products tested were injected subcutaneously into the thorax. The time corresponding to the administration of the product is noted as T 0. Different groups were defined according to the number of days between aspirin administration and the laser-induced thrombosis. To induce thrombosis, the small intestine was exteriorized through a median laparotomy and the mesentery was spread flat on a glass plate mounted on the microscope table. Investigation was performed in vessels of the same diameter (15–25 μm) of the flat-free portion of the mesentery. At least seven rats were studied in each group. Test and control animals were selected at random.
Two days after administration, aspirin still demonstrated potent antithrombotic activities. In fact, compared with the control group, aspirin administered at 100 mg/kg 2 days before thrombosis induction decreased the NE and reduced the DE (p < 0.05). An opposite effect of the administration of aspirin (100 mg/kg) 8 days and, to lesser extent, 10 days before thrombosis induction was observed, since the NE was increased and the DE was prolonged (p < 0.05) [26]. There was a potent antithrombotic effect 2 days after injection (T 0 + 2 days). Thrombus formation was significantly increased at T 0 + 8 days and T 0 + 10 days, expressing in healthy rats results similar to the observational epidemiological data described above [6]. This experiment clearly demonstrated the antithrombotic effect of aspirin 2 days after injection and showed that a prothrombotic state can be observed several days after a single aspirin dose in healthy rats for a given period of time. A rebound effect could not be ruled out in this experiment, but this effect was observed in normal rats and was similar to the prothrombotic effect of ULDA produced 1 h after aspirin injection.
This investigation demonstrated three phases of ASA action according to the delay between its administration and the laser-induced thrombosis. First, aspirin showed a potent antithrombotic effect that disappeared 3 days after administration. The difference between the control group and the ASA group 2 days after administration reached the level of significance (p < 0.05). In the second phase, aspirin did not show any effect between 3 and 7 days after administration. In contrast, 8 and 10 days after administration, aspirin increased the NE and the DE, testifying to its prothrombotic properties.
The administration of aspirin 100 mg/kg can first prevent the thrombus formation induced by endothelial cell damage. However, a long time after the treatment, it increases the risk of thromboembolic complications. Therefore, the effect of aspirin may depend on the drug concentration in the microvasculature, its pharmacokinetics, and its availability as ASA at the vascular level. The effect may be related to an ‘effect curve,’ i.e., small concentrations of aspirin causing large changes in effect on a very steep concentration gradient. This would be in accordance with the prolonged presence of the drug at least up to 6 h after oral administration, despite the transient duration of its effects. Another mechanism could be the generation or the activation of the synthesis of an active metabolite with prothrombotic activity or the inhibition of the synthesis of an agent with antithrombotic activity by ultra-low concentrations in plasma. Such an effect has been previously reported by Czevionke et al. [27]. Thus, inhibition of vascular COX by administration of aspirin could lead to the generation of platelet activating factor (PAF), which is a potent stimulus for the activation of platelets [28] and could cause subsequent endothelial injury.
The mechanism of this action is unknown, but it could be due to the accumulation of very small doses of aspirin in the microvasculature or to other as yet unknown actions. This prothrombotic activity of aspirin 8 days after a single-dose administration may explain the thromboembolic phenomenon observed in patients when they stop aspirin treatment. These findings should be taken into consideration if the end of aspirin therapy is planned, and suggest the need for further studies.
3 Possible Mechanisms Explaining the Opposite Effects of Aspirin on Platelet Activity Depending on the Dose Administered
3.1 Effects of Administration of Aspirin Alone or Associated with Specific Cyclooxygenase-1 (COX-1) or COX-2 Inhibitors
We first studied the effects of aspirin when administered in different doses alone or in association with specific COX-1 (SC-560) or COX-2 (NS-398) inhibitors tested at different doses (2.5, 5, 7.5, and 10 mg/kg) or their association at 10 mg/kg in the LITM. This study aimed to clarify the effect of the lower end of the aspirin dose-response curve, the differences observed with higher doses, the possible mechanism involved and its clinical implications.
3.1.1 Drug Levels Tested
The amounts of 1 mg/mL and 100 mg/mL were obtained by dilution of a solution of acetylsalicylate. Other aspirin dilutions were prepared as follows: 1 g of pure, finely powdered aspirin was suspended in 99 mL of alcohol (70°). After being vigorously shaken, 1 mL of this dilution was then mixed with 99 mL of distilled water and vigorously shaken (dilution 1). The latter process was repeated until the required dilutions were obtained: 4-fold (dilution 5), 8-fold (dilution 9), and 14-fold (dilution 15). Alcohol and sterilized water according to the abovementioned procedures without adding aspirin were used as placebo for the dilutions 1, 5, 9, and 15. Sterilized water for injectable preparations was used as placebo for groups receiving 100 mg/kg or 1 mg/kg of aspirin. Aspirin or the corresponding placebo was administered subcutaneously to rats at a final volume of 1 mL/kg bodyweight. The different placebos were used to avoid interference due to the different kinds of aspirin preparation used.
Selective COX-1 (SC-560) and COX-2 (NS-398) inhibitors were suspended in carboxy-methyl-cellulose (CMC) 0.5 g/L at a final volume of 1 mL/kg bodyweight. The CMC solution without the inhibitors was used as placebo. Selective COX inhibitors were administered per os to rats at doses of 2.5, 5, 7.5, or 10 mg/kg when used alone and at a dose of 10 mg/kg when both were administered simultaneously [29].
3.1.2 Distribution of Groups
This study comprised five experiments as follows:
Experiment 1 Action of aspirin 100 mg/kg alone or associated with specific COX-1 (SC-560) or COX-2 (NS-398) inhibitors tested to increasing amounts (2.5, 5, 7.5, and 10 mg/kg) or their association at 10 mg/kg on thrombosis and hemostasis.
Experiment 2 Action of aspirin 1 mg/kg alone or associated with specific COX-1 (SC-560) or COX-2 (NS-398) inhibitors tested to increasing amounts (2.5, 5, 7.5, and 10 mg/kg) or their association at 10 mg/kg on thrombosis and hemostasis.
Experiment 3 Action of aspirin in dilution 5, 1 mL/kg bodyweight alone or associated with specific COX-1 (SC-560) or COX-2 (NS-398) inhibitors tested to increasing amounts (2.5, 5, 7.5, and 10 mg/kg) or their association at 10 mg/kg on thrombosis and hemostasis.
Experiment 4 Action of aspirin dilution 9, 1 mL/kg bodyweight alone or associated with specific COX-1 (SC-560) or COX-2 (NS-398) inhibitors tested to increasing amounts (2.5, 5, 7.5, and 10 mg/kg) or their association at 10 mg/kg on thrombosis and hemostasis.
Experiment 5 Action of aspirin dilution 15, 1 mL/kg bodyweight alone or associated with specific COX-1 (SC-560) or COX-2 (NS-398) inhibitors tested to increasing amounts (2.5, 5, 7.5, and 10 mg/kg) or their association at 10 mg/kg on thrombosis and hemostasis.
Experimental groups: 
Results showed that aspirin at 100 mg/kg decreased thrombus formation. This effect of the higher dose of aspirin was similar to the effect of selectively inhibiting COX-1. Interestingly, the addition of selective COX-1 inhibition with SC-560 or 100 mg/kg of aspirin had the same effect. These effects were not synergistic, as both are thought to function through the same mechanism on thrombus production [30]. Selective COX-2 inhibition did not modify the antithrombotic effect of aspirin at the highest dose, probably owing to the vasoconstriction involved in COX-2 inhibition.
Aspirin at 1 mg/kg has a less marked effect by mildly decreasing NE. A significant increase in NE and DE occurred with ULDA, confirming previous data [24]. After selective COX inhibition at different doses, ULDA gave a radically different result from that of aspirin at 100 mg/kg. Administration of ULDA to rats previously treated with the selective COX-2 inhibitor produced no further effect. These observations led us to conclude that high dilutions of aspirin have a COX-2 inhibiting effect [30]. This prothrombotic reaction is not modified by COX-2 inhibition, although it is decreased somewhat by the antithrombotic effect of selective COX-1 inhibition. This decrease in the prothrombotic effect of ULDA could be due to a partially protective effect in the platelets against COX-1 inhibition. Another possible explanation is the prothrombotic effect of COX-2 inhibition counterbalanced to some extent by the decrease in platelet TXA2 production observed after selective COX-1 inhibition. A group treated with both COX-1 and COX-2 inhibitors plus the dilutions of aspirin underwent the same detrimental effect.
Aspirin modified thrombi production by decreasing the number with higher doses and increasing it with ultra-low dose. The lowest dilution studied had the effect of protecting COX-1 against inhibition or directly inhibiting COX-2, leading to a strong prothrombotic state.
3.2 Effects of Aspirin on Genetically Modified Homozygous Male Mice
To confirm these results, we designed an experiment using 72 genetically modified male homozygous knockout mice without COX-1 (COX-1 −/−) and 72 knockout mice lacking COX-2 (COX-2 −/−) to evaluate primary hemostasis. Aspirin doses used were 100 mg/kg bodyweight, 1 mg/kg bodyweight and aspirin 1/100 dilutions number 5 (dilution 5), 9 (dilution 9) and 15 (dilution 15), which were obtained by successive 1/100 dilution as described above. Sterilized water was used as placebo. All drugs were injected subcutaneously at a final volume of 1 mL/kg bodyweight.
In this study, we used healthy mice and the male homozygous COX-1 −/− and COX-2 −/− mice (knockout).
Distribution of groups
COX-1 −/− or COX-2 −/− knockout mice were distributed in six groups (n = 12 per group) respectively:
Group 1: Placebo (sterilized water)
Group 2: Aspirin 100 mg/kg
Group 3: Aspirin 1 mg/kg
Group 4: Aspirin dilution 5
Group 5: Aspirin dilution 9
Group 6: Aspirin dilution 15
The NE and DE in COX-1-deficient mice were clearly decreased with placebo when compared with COX-2-deficient mice, highlighting the importance of COX-1-generated TXA2 in platelets and indicating the presence of an antithrombotic mechanism different to COX-1 inhibition at this dose. This model of COX-1 −/− mouse is known to produce alterations in platelet aggregation. Explanations for this effect may include compensation in COX-1 and COX-2 activity or an interaction between COX and NO synthase, as suggested by Skill et al. [31], or an effect of aspirin outside the mechanism of COX inhibition, e.g., increased NO synthesis by the endothelial cell, as suggested by Taubert et al. [32]. Although intermediate doses did not show any significant effects, there was a clear antithrombotic effect of high-dose aspirin in COX-2-deficient mice and a prothrombotic effect with the lowest dilution in COX-1-deficient mice. These results clearly confirm that although high-dose aspirin (100 mg/kg) exerts its primary antithrombotic effect through COX-1 inhibition, dilution 15 acts via COX-2 inhibition to induce thrombosis.
In COX-1-deficient mice, aspirin had a mild antithrombotic effect at a high dose (100 mg/kg) and a strong prothrombotic effect at dilution 15. Both effects were independent of COX-1 activity. Dilution 15 had no effect in COX-2-deficient mice. The effect observed with the lowest dose (dilution 15) suggests that the prothrombotic effect after aspirin withdrawal is directly due to residual amounts of aspirin rather than because of a rebound effect, and that the complications observed after aspirin discontinuation may arise from this, as yet, unrecognized effect.
4 Discussion
Drug withdrawal effects are usually disregarded in pharmacology. The recent demonstration of increased mortality after discontinuation of aspirin prophylaxis, especially in the first 5–7 days after stopping treatment, indicates the need to elucidate the mechanisms underlying this effect. Rebound effects may explain a part of the mechanisms involved. The possibility of medication discontinuation syndromes arising on cessation of drugs administered for a long time requires more research and attention to reduce the potential harm that discontinuation may cause.
When a drug is stopped, the underlying state can recur. The withdrawal syndromes seem to have different biological bases. Some examples of cardiovascular disease exacerbation, with some fatalities, after cessation of specific cardiovascular drugs are described below.
Some of the first biological and clinical studies demonstrated that urinary excretion of both 6-keto-prostaglandin F1α and TXB2 were elevated 2 weeks after aspirin was stopped [33]. A few years later, it was demonstrated that although platelets are inhibited by a single dose of aspirin, fibrinogen binding to activated platelets was increased 6 days after the dose compared with baseline, as was arachidonic acid-induced platelet aggregation [34]. The effect of aspirin discontinuation was also demonstrated on fibrin–fibrinogen interaction [35]. Sudden discontinuation of long-term aspirin therapy seemed to increase the risk of acute coronary thrombosis [12]. As studies continue to focus on this problem, the biological and clinical evidence for a potential aspirin discontinuation effect increasing the risk of thrombotic events has accrued. The rebound effect caused by discontinuation of aspirin has been reviewed with the severe adverse clinical consequences described and the risk of enhanced ischemic stroke [36].
The abovementioned results showed that ULDA alone exerts prothrombotic activities and increases the NE and the DE. Specific COX-1 inhibition has antithrombotic effects in rats. On the other hand, specific COX-2 inhibition has prothrombotic activities that are not enhanced by the administration of ULDA. High-dose aspirin in COX-1-deficient mice has a mild effect by decreasing thrombosis and a strong prothrombotic effect at dilution 15. Both effects are independent of COX-1 activity. Dilution 15 has no effect in COX-2-deficient mice, an effect that might be directly due to residual amounts of aspirin rather than to a rebound effect. The complications observed after aspirin discontinuation may arise from this, as yet, unrecognized effect. These results suggest that the prothrombotic effects of ULDA may occur via a COX-2 pathway rather than via a COX-1 route. This would explain the reported thromboembolic complications observed several days after aspirin withdrawal, a phenomenon usually interpreted as a rebound effect. However, this study suggests that this thromboembolic complication is more likely a direct effect of ULDA via COX-2 inhibition.
5 Conclusions
These experiments demonstrate that the withdrawal of aspirin can induce a prothrombotic state several days later. This prothrombotic state is reproducible after the administration of ULDA, which can induce the same prothrombotic state 1 hour after administration. The mechanism of this effect includes COX-2 inhibition. Further work is required to prevent this negative effect, which is certainly not specific to aspirin. When they are stopped, other drugs might promote a similar secondary effect at ultra-low doses.
These findings could have important implications for public health and also highlight the importance of extended dose-response studies for pharmaceutical compounds beyond toxic and therapeutic doses. Moreover, they show that the presence of an inactive dose threshold does not imply inactivity at lower concentrations, which might be the unexpected source of side effects.
Further research is warranted to better understand drug withdrawal effects. This will hopefully make it possible to safely withdraw aspirin and possibly other drugs.
References
Chen ZM, Jiang LX, Chen YP, Xie JX, Pan HC, Peto R, Collins R, Liu LS; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366(9497):1607–21.
Bhatt DL, Fox KA, Hacke W, Berger PB, Black HR, Boden WE, Cacoub P, Cohen EA, Creager MA, Easton JD, Flather MD, Haffner SM, Hamm CW, Hankey GJ, Johnston SC, Mak KH, Mas JL, Montalescot G, Pearson TA, Steg PG, Steinhubl SR, Weber MA, Brennan DM, Fabry-Ribaudo L, Booth J, Topol EJ; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med. 2006;354(16):1706–17.
Sibon I, Orgogozo JM. Antiplatelet drug discontinuation is a risk factor for ischemic stroke. Neurology. 2004;62(7):1187–9.
Maulaz AB, Bezerra DC, Michel P, Bogousslavsky J. Effect of discontinuing aspirin therapy on the risk of brain ischemic stroke. Arch Neurol. 2005;62(8):1217–20.
Armstrong MJ, Schneck MJ, Biller J. Discontinuation of perioperative antiplatelet and anticoagulant therapy in stroke patients. Neurol Clin. 2006;24(4):607–30.
Fischer LM, Schlienger RG, Matter CM, Jick H, Meier CR. Discontinuation of nonsteroidal anti-inflammatory drug therapy and risk of acute myocardial infarction. Arch Intern Med. 2004;164(22):2472–6.
Ferrari E, Benhamou M, Cerboni P, Marcel B. Coronary syndromes following aspirin withdrawal: a special risk for late stent thrombosis. J Am Coll Cardiol. 2005;45(3):456–9.
Burger W, Chemnitius JM, Kneissl GD, Rucker G. Low-dose aspirin for secondary cardiovascular prevention—cardiovascular risks after its perioperative withdrawal versus bleeding risks with its continuation—review and meta-analysis. J Intern Med. 2005;257(5):399–414.
Collet JP, Montalescot G, Blanchet B, Tanguy ML, Golmard JL, Choussat R, Beygui F, Payot L, Vignolles N, Metzger JP, Thomas D. Impact of prior use or recent withdrawal of oral antiplatelet agents on acute coronary syndromes. Circulation. 2004;110(16):2361–7.
Ho PM, Spertus JA, Masoudi FA, Reid KJ, Peterson ED, Magid DJ, Krumholz HM, Rumsfeld JS. Impact of medication therapy discontinuation on mortality after myocardial infarction. Arch Intern Med. 2006;166(17):1842–7.
Biondi-Zoccai GG, Lotrionte M, Agostoni P, Abbate A, Fusaro M, Burzotta F, Testa L, Sheiban I, Sangiorgi G. A systematic review and meta-analysis on the hazards of discontinuing or not adhering to aspirin among 50,279 patients at risk for coronary artery disease. Eur Heart J. 2006;27(22):2667–74.
Collet JP, Himbet F, Steg PG. Myocardial infarction after aspirin cessation in stable coronary artery disease patients. Int J Cardiol. 2000;76(2–3):257–8.
Albaladejo P, Geeraerts T, Francis F, Castier Y, Leseche G, Marty J. Aspirin withdrawal and acute lower limb ischemia. Anesth Analg. 2004;99(2):440–3.
Eisenberg MJ, Richard PR, Libersan D, Filion KB. Safety of short-term discontinuation antiplatelet therapy in patients with drug-eluting stents. Circulation. 2009;119(12):1634–42.
Fitzgerald GA. Parsing an enigma: the pharmacodynamics of aspirin resistance. Lancet. 2003;361:542–4.
Awtry EH, Loscalzo J. Aspirin. Circulation. 2000;101:1206–18.
Wu KK. Aspirin and salicylate: an old remedy with a new twist. Circulation. 2000;102:2022–3.
Gurfinkel EP, Altman R, Scazziota A, Heguilen R, Mautner B. Fast platelet suppression by lysine acetylsalicylate in chronic stable coronary patients. Potential clinical impact over regular aspirin for coronary syndromes. Clin Cardiol. 2000;23(9):697–700.
Patrignani P, Filabozi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin unhealthy subjects. J Clin Invet. 1982;69:1366–72.
Reilly IAG, FitGerald GA. Inhibition of thromboxane formation in vivo and ex vivo. Blood. 1987;69:180–6.
Hoogendijk EMG, ten Cate JW. Aspirin and platelets. Lancet. 1980;1(8159):93–4.
Jakubowski JA, Stamfer MJ, Vaillancourt R, Deykin D. Cumulative ant platelet effect of low-dose enteric coated aspirin. Br J Haematol. 1982;60:635–42.
Vesvres MH, Doutremepuich F, Lalanne MC, Doutremepuich C. Effects of aspirin on embolization in an arterial model of laser-induced thrombus formation. Haemostasis. 1993;23(1):8–12.
Doutremepuich C, Aguejouf O, Belon P. Effects of ultra-low-dose aspirin on embolization in a model of laser-induced thrombus formation. Semin Thromb Hemost. 1996;22(Suppl 1):67–70.
Aguejouf O, Eizayaga F, Desplat V, Belon P, Doutremepuich C. Prothrombotic and hemorrhagic effects of aspirin. Clin Appl Thromb Hemost. 2009;15(5):523–8.
Aguejouf O, Belougne-Malfatti E, Doutremepuich F, Belon P, Doutremepuich C. Thromboembolic complications several days after a single-dose administration of aspirin. Thromb Res. 1998;89(3):123–7.
Czevionke RL, Hoak M, Fry GL. Effect of aspirin on non thrombin-induced adherence of platelets to cultured vessel wall. J Clin Invest. 1978;62:847–56.
Mc Manus LM, Hanahan, DJ, Pinckard RN. Human platelet stimulation by acetyl glyceryl ether phosphorylcholine. J Clin Invest. 1981;67:903–6.
Doutremepuich C, Aguejouf O, Desplat V, Eizayaga FX. Paradoxical thrombotic effects of aspirin: experimental study on 1000 animals. Cardiovasc Hematol Disord Drug Targets. 2010;10(2):103–10.
Doutremepuich C, Aguejouf O, Eizayaga FX, Desplat V. Reverse effect of aspirin: is the pro-thrombotic effect after aspirin discontinuation mediated by cycloxygenase 2 inhibition? Pathophysiol Haemost Thromb. 2007;36(1):40–4.
Skill NJ, Theodorakis NG, Wang YN, Wu JM, Redmond EM, Sitzmann JV. Role of cyclooxygenase isoforms in prostacyclin biosynthesis and murine prehepatic portal hypertension. Am J Physiol Gastrointest Liver Physiol. 2008;295(5):G953–64.
Taubert D, Berkels R, Grosser N, Schröder H, Gründemann D, Schömig E. Aspirin induces nitric oxide release from vascular endothelium: a novel mechanism of action. Br J Pharmacol. 2004;143(1):159–65.
Vial JH, McLeod LJ, Roberts MS. Rebound elevation in urinary thromboxane B2 and 6-keto-PGF1 alpha excretion after aspirin withdrawal. Adv Prostaglandin Thromboxane Leukot Res. 1991;21A:157–60.
Mousa SA, Forsythe MS, Bozarth JM, Reilly TM. Effect of single oral dose of aspirin on human platelet function and plasma plasminogen activator inhibitor-1. Cardiology. 1993;83:367–73.
Fatah K, Beving H, Albâge A, Ivert T, Blombäck M. Acetylsalicylic acid may protect the patient by increasing fibrin gel porosity. Is withdrawing of treatment harmful to the patient? Eur Heart J. 1996;17:1362–6.
Lordkipanidzé M, Diodati JG, Pharand C. Possibility of a rebound phenomenon following antiplatelet therapy withdrawal: a look at the clinical and pharmacological evidence. Pharmacol Ther. 2009;123:178–86.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Doutremepuich, C., Aguejouf, O., Desplat, V. et al. Aspirin Discontinuation Syndromes: Clinical Implications of Basic Research Studies. Am J Cardiovasc Drugs 13, 377–384 (2013). https://doi.org/10.1007/s40256-013-0044-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-013-0044-1