
Abstract
Objectives
Diabetic neuropathic pain (DNP) is a debilitating symptom of diabetic neuropathy which seriously impairs patient’s quality of life. Currently, there is no specific therapy for DNP except for duloxetine and gabapentin that show limited utility in alleviating DNP. The present review aims to discuss the central role of protein kinases in the pathogenesis of DNP and their therapeutic modulation.
Methods
Scopus, PubMed, and Google scholar were searched up to January 2022 to find relevant studies with English language in which the roles of proteins kinases in DNP were examined.
Results
DNP is associated with hyperactivity in pain sensory neurons and therapies aim to specifically suppress redundant discharges in these neurons without affecting the activity of other sensory and motor neurons. Transient receptor potential vanilloid 1 (TRPV1) and purinergic 2 × 7 receptors (P2 × 7R) are two receptor channels, highly expressed in pain sensory neurons and their blockade produces remarkable analgesic effects in DNP. The activities of receptor channels are mainly regulated by the protein kinases whose modulation provides remarkable analgesic effects in DNP models.
Conclusion
Capsaicin, TRPV1 modulator, is the only agent successfully examined in clinical trials with promising effects in patients with DNP. Current data suggest that blocking calcium calmodulin dependent protein kinase II (CaMKII) is superior to other approaches, considering its pivotal role in regulating the pain neuron potentials. By this means, DNP alleviation is achievable without affecting the activity of other sensory or motor neurons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes is the largest global epidemic of the current century, affecting 425 million people worldwide, and diabetic neuropathy is one of its common complications that occurs in nearly half of all diabetic patients. Diabetic neuropathy is caused by damage to the peripheral (both motor and sensory) and autonomic neurons [1], and the most common form of diabetic neuropathy is distal symmetric polyneuropathy where the extremities (hands, toes, and feet) are commonly affected. It manifests as motor weakness and sensory symptoms including paresthesia (abnormal painless sensations like numbness, tingling, itching), dysesthesia (abnormal painful sensations like pricking, burning, ice-old), allodynia (pain evoked by normally nonpainful stimuli like touching, running of water), and hyperalgesia (exaggerated pain sensation in response to mild painful stimuli) [2, 3]. Diabetic neuropathic pain (DNP) is a general term that encompasses dysesthesia, allodynia, and hyperalgesia which markedly impairs patients’ life quality, affects sleep, work, self-esteem, and social relations, and results in social abstentions and depression [4]. Generally, one quarter of all diabetic patients suffer from DNP [5].
In DNP, a physical damage to pain neurons is not the major problem, rather pain sensory neurons become hyperactive and discharge frequently in this condition. Pain sense is transmitted to the central nervous system by dorsal root ganglia (DRG) and through un-myelinated C- and thinly myelinated A-fibers, and it is now known that the sensitization and hyperactivity of these neurons are the mainstay of DNP [6]. In the last two decades, efforts have been made to elucidate the molecular basis underlying the neuronal hyperactivity in DNP in order to tailor specific and effective treatment methods for the condition [7]. A-fiber neurons in the DRG are large-diameter neurons with thinly myelinated axons that convey the pain sensation, and have pivotal roles in the induction of allodynia in neuropathic pains [8].
A protein kinase is a kinase enzyme that enacts protein phosphorylation by covalently adding a phosphate group from adenosine triphosphate (ATP) to an amino acid residue in a protein. Protein phosphorylation is a reversible post-translational modification which alters the protein conformation, and results in either target activation or deactivation. Protein kinases are categorized into two main types: 1- serine/threonine kinases, that target the hydroxyl groups of serine or threonine residues in the downstream proteins, and 2- tyrosine kinases, which phosphorylate the tyrosine residues in their targets [9]. Kinases are extremely crucial for cell biology as ∼30% of all human proteins could be modified by kinases; and therefore, these enzymes regulate the majority of cellular functions [10].
Molecular pathogenesis of DNP: the central role of receptor channels
Receptor channels, also called ligand-gated ion channels, are a set of transmembrane multimeric proteins located on the neurons, that open to allow the passage of diverse ions like Na+, K+, Ca2+, and/or Cl− in response to the binding of a chemical messenger (i.e. a ligand), such as a neurotransmitter [11]. These channels act to regulate or control the function of neurons, and depending on the ion that allow in or out of the cell, they stimulate or suppress the neuronal function. For instance, increased load of Cl− ions inside neurons opposes the induction of action potential, making the neurons less excitable; conversely, increased load of positively charged ions such as Na+ and Ca+ decreases the stimulatory threshold and renders the neurons hyperactive [12, 13]. Under diabetic conditions, the expression of receptor channels as well as the production of neurotransmitters are altered which affects the activity of neurons. Therefore, recognizing the pathophysiology of receptor channels in diabetes will elucidate the nature of neuronal derangements seen in these patients and helps in tailoring more targeted therapies [14].
The transient receptor potential vanilloid 1 (TRPV1) is a ligand-gated nonselective cation channel that specifically localizes to C- and A-fiber (nociceptive) sensory neurons [15]. TRPV1 activation allows for the inflow of Na+ and Ca2+ ions, initiating nerve depolarization. This key role of TRPV1 in nociception has been corroborated by the finding that TRPV1-knockout mice had decreased pain response following thermal hyperalgesia [16]. Kamei et al., were the first scholars to show that intrathecal injection of anti-TRPV1 serum to diabetic mice alleviated thermal allodynia [17]. Moreover, substance P is the main neurotransmitter in afferent pain fibers, whose expression is greatly increased in neuropathic pains, and it has been evidenced that capsaicin, a TRPV1 activator, triggers these neurons to eventually deplete their substance P reserves, causing pain attenuation both in diabetic mice and in human patients; in fact, prolonged exposure to capsaicin desensitizes the pain nociceptors [18]. Capsaicin 8% patch causes no neurologic adverse effects in diabetic patients [19] and attenuates pain two weeks after the treatment onset [20]. Similarly, capsazepine, a TRPV1 antagonist, effectively alleviated pain in rat, mouse, and guinea pig models of capsaicin-induced, inflammatory, and neuropathic pains [21].
The transient receptor potential M8 (TRPM8), formerly called menthol and cold receptor 1 (CMR1), is the other non-selective ion channel present on pain sensory neurons. Its role in neuropathic pains is less investigated compared to TRPV1. TRPM8 contributes to cold sensation and is evoked by temperatures lower than 25°C, as well as by cooling agents like menthol and icilin [22]. TRPM8 activation results in the influx of Na+ and Ca2+ with the consequent membrane depolarization [23]. phosphatidylinositol 4,5-bisphosphate (PIP2) is the pivotal regulator of TRPM8 channels, as exogenous PIP2 activates them in DRG neurons in vitro; conversely, inhibition of phosphoinositide (PI) 4-kinase, the enzyme responsible for PIP2 synthesis, by either wortmannin or phenylarsine oxide (PAO) down-regulated the TRPM8 activity [24]. Total protein levels as well as phosphorylated forms and the activity of TRPM8 channels are increased in the DRG neurons of diabetic mice, and both protein kinase A and protein kinase C contribute significantly to the enhanced activities of these channels. Moreover. Specific inhibition of TRPM8 channels significantly attenuates pain indices in diabetic mice [25].
Acid sensing ion channel 1 (ASIC1) is a proton-gated cation channel which allows for the influx of Na+ ions, and is widely expressed in nociceptive neurons [26]. It has been shown that ASIC1 protein levels are increased in the DRG neurons of diabetic rats [27]. However, it is not yet known that whether the activity of ASIC1 is altered under diabetic circumstances or not, and if ASIC1 stimulation or blockade has any therapeutic utility in DNP. Therefore, more thorough investigations are needed to answer these questions.
Orphan G protein–coupled receptor 177 (GPR177) is a seven-transmembrane protein and is principally expressed in A-fiber DRG neurons that regulates the secretion of wingless-type mammary tumor virus integration (Wnt) ligands [28]. Despite not being an ion channel, it is discussed here because GRP177 is closely related to TRPV1 function. GRP177 is specifically up-regulated in A-fiber DRG neurons and exclusively secretes Wnt5a from these neurons in diabetic mice. Wnt5a secretions is essential for allodynia and hyperalgesia as its blockade abrogates these symptoms in diabetic mice. Wnt5a increases intracellular levels of Ca2+ and in turn activates the TRPV1 channels [29].
Purinergic 2 × 7 receptors (P2 × 7R) are non-selective cation channels that are principally expressed in the DRG neurons. These receptor channels are mainly activated by the extracellular adenosine triphosphate (ATP). In neuropathic conditions, the ATP released from the injured neurons and glial cells stimulate P2 × 7Rs on the intact pain neurons [30]. P2 × 7R is also present on the glial cells of DRG whose stimulation leads to the release of inflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin 1β (IL-1β), and plasminogen which aggravate the neuropathic pain [31]. It was detected in diabetic rats that the expression of P2 × 7R in DRG neurons is increased, and intrathecal administration of A438079, the P2 × 7R antagonist, improved allodynia and pain indices, and simultaneously decreased protein levels of TRPV1. Moreover, it decreased IL-1β levels (a pro-inflammatory cytokine) and elevated IL-10 levels (an anti-inflammatory cytokine). It was found that P2 × 7R inhibition reduced the expression of TRPV1 by down-regulating the MAPK signaling pathway [32]. Another study conducted on diabetic rats reported that either silencing P2 × 7R or inhibiting p38 improved allodynia, attenuated both TRPV1 and PKC expressions, and simultaneously decreased IL-1β levels by suppressing NF-kB activity [33].
Cannabinoid 2 receptor (CB2R) is expressed on the DRG neurons whose activation produces analgesia. Intrathecal administration of AM1241 and AM1710, the CB2R agonists, in rats with sciatic nerve injury alleviated allodynia and suppressed inflammation in the dorsal horn of the spinal cord [34]. It should be underlined that the increased levels of neuroimmune chemokine, C–C class chemokine-2 (CCL2), also called monocyte chemo-attractant protein-1 (MCP-1), in the dorsal horn of spinal cord increases the expression of TRPV1 on DRG neurons [35]. However, it has been shown that AM1710 (CB2R agonist) attenuates allodynia both independently of TRPV1 and by down-regulating TRPV1 expression on DRG neurons [36].
Protein kinase C (PKC)
PKC is a serine/threonine kinase which controls the function of other proteins by phosphorylation. PKC is mainly activated by diacylglycerol (DAG), a second messenger lipid, which is generated by hydrolysis from the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) by the function of membrane-anchored enzyme phospholipase C (PLC). Inositol trisphosphate (IP3) is the other product of PLC which enters the cytosol and induces the release of calcium ions from the smooth endoplasmic reticulum, whereas DAG does not diffuse in to the cytoplasm, remains in the plasma membrane due to its hydrophobic properties and directly activates PKC. Moreover, PKC can be activated by calcium ions (Ca2+) [37].
PKC is expressed in high concentrations in neuronal tissues and is involved in a wide array of neuronal functions [38]. PKC activates TRPV1 by phosphorylation. It has been demonstrated that TRPV1 could solely be activated by PKC, even in the absence of TRPV1 agonist. TRPV1 activation was augmented by tetradecanoylphorbol acetate (TPA), a PKC activator; and its activity was reduced by bisindolylmaleimide (BIM), a PKC inhibitor [39]. These findings were confirmed in vivo by two independent studies, as it was shown that both phosphorylated protein levels and the activity of DRG neurons were increased in rats with diabetes; and these effects were enhanced after administering a PKC activator, and were abrogated after injecting a PKC inhibitor [40, 41].
Advanced glycation end products (AGEs) are formed in excess quantities in diabetes by non-enzymatic reaction of glucose with proteins. AGEs contribute to the pathogenesis of diabetic complications by activating the receptor for advanced glycation end products (RAGEs), expressed on various cell types [42]. High glucose concentrations evoke the TRPV1 activity in mice DRG neurons in vitro while the cell viability was not affected in the high glucose medium. Interestingly, RAGE-knockout DRG neuros demonstrated no hyperactivity in high glucose medium; likewise, wild-type DRG neurons exposed to antioxidants α-lipoic acid plus catalase showed no hyperactivity in the same medium. Finally, inhibition of either PKC or Drc kinase abrogated RAGE-mediated hyperactivity in DRG neurons, suggesting that RAGE contributes to the hyperactivity of TRPV1 channels by activating PKC and Src kinase [43].
Protein kinase (PKA)
PKA, also called cAMP-dependent protein kinase, is a kinase which is regulated by cyclic AMP (cAMP). Extracellular molecules such as serotonin, prostaglandins, and epinephrine modulate nociception by first binding to a G protein–coupled receptor (GPCR) on the neuron. When activated, a conformational change is induced in the receptor that is transmitted to an attached intracellular heterotrimeric G protein complex through protein domain dynamics. The Gs alpha subunit of the stimulated G protein complex exchanges GDP for GTP in a reaction catalyzed by the GPCR and is released from the complex. The activated Gs alpha subunit binds to and activates an enzyme called adenylyl cyclase, which, in turn, catalyzes the conversion of ATP into cAMP, directly increasing the cAMP level.
It has been shown that the injection of membrane permeable cAMP or adenylyl cyclase activators lower the nociceptive threshold and lead to hyperalgesia in experimental models [44]. Mechanistically, PKA phosphorylates the hyperpolarization activated cation channels (HCN) on the nociceptive neurons to allow in more positively charged ions, leading to frequent nerve activation [45]. PKA also induces the activity of voltage-gated sodium channels on the pain sensory nerves which augments their excitability [46]. It has been shown that dexmedetomidine relieves hyperalgesia induced by brachial plexus root avulsion by suppressing PKA [47]. PKA is also able to directly phosphorylate TRPV1 channels. It has been shown that prostaglandin E2 (PGE2) induces hyperalgesia by increasing intracellular c-AMP levels (PKA activation); conversely, the µ-opioid agonists produce analgesic effects by decreasing intracellular c-AMP levels (PKA inhibition) [48].
Protein kinase B (PKB)
PKB, also commonly called Akt, is another serine/threonine protein kinase that has multiple roles in various cellular functions including cell survival, proliferation, and apoptosis [49]. unlike other protein kinases, PKB is part of the PI3K/Akt/mTOR signaling pathway; and it is activated by phosphoinositide 3-kinase (PI3K), and then activates the downstream kinase, mammalian target of rapamycin (mTOR) which is the main effector protein kinase of this pathway [50]. mTOR activation downregulates autophagy in neuronal tissues, limiting the cells adaptive properties and contributes to the pathogenesis of neuropathic pains [51]. Impaired autophagy is associated with increased activity of pain sensory fibers in rats, exacerbating allodynia and hyperalgesia indices [52]. It has been shown that in the DRG neurons of diabetic rats, the phosphorylated levels of all PI3K, PKB, and mTOR are increased, suggesting the activation of this pathway; and the levels of belcin-1 and LC3-II, the autophagy-related proteins, are decreased, suggesting the inhibition of autophagy. It was found that LY294002, the PI3K inhibitor, reversed the above-mentioned indices and significantly attenuated pain perception in diabetic rats [53]. Whether PI3K/Akt/mTOR activation in the pain sensory neurons of diabetic rats affects TRPV1 channels or not has not been investigated yet.
Calcium/calmodulin-dependent protein kinase (CaMK)
CaMK is a serine/threonine kinase activated by elevations in the intracellular levels of Ca2+ and calmodulin. As a calcium-binding protein, calmodulin is being targeted by the secondary messenger Ca2+. Ca2+/calmodulin complex functions as part of a calcium signal transduction pathway by activating CaMK. Activated CAMK phosphorylates a broad spectrum of membrane-bound to transcription factor proteins, and thereby, regulates many cellular functions including cell division, proliferation, and programmed cell death [54]. CaMKII is the main isoform present in neuronal cells [55], and therefore, in the following lines the role of CaMKII will be discussed in DNP. In basal state, CaMKII is phosphorylated at Thr305. When Ca2+ binds CaMKII, it gains an auto-phosphorylating property to be self-phosphorylated on Thr286 which is the active form of the enzyme [56]. Auto-phosphorylated CaMKII does not need further stimulation by calcium and it can maintain its activity independent of further stimuli, providing a function of molecular memory [57].
Nearly a decade ago, two independent experimental studies on rat models of diabetes demonstrated increased protein expression and phosphorylation of CaMKII in DRG neurons. According to these reports, the α isoform of CaMKII was the predominant form, and that the increased CaMKIIα activity was associated with pain-related behavior in rats [58, 59]. Increased activity of CaMKIIα in trigeminal nerves of diabetic rats has been noticed [60]. Moreover, myristoylated autocamtide-2-inhibitory peptide (AIP) produces analgesia in rats with sciatic nerve mononeuropathy by inhibiting the CaMKII [61]. Both total protein and phosphorylated forms of CaMKII are increased in the DRG neurons of diabetic rats, and KN-93, the CaMKII inhibitor, alleviates hyperalgesia, in addition to reducing phosphorylated CaMKII and P2 × 3R levels [62].
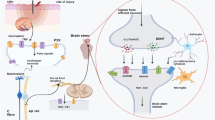
N-methyl-D-aspartate receptor (NMDAR) is a receptor-channel activated by the salient excitatory neurotransmitter glutamate, and is widely expressed on pain sensory neurons [63]. Upon activation, it allows for the inflow of Ca2+ ions. It was first demonstrated by Matsumura and colleagues that NMDAR-knockout mice had decreased allodynia in sciatic neuropathic pain. Moreover, in NMDAR-knockdown mice, the concentration of intracellular Ca2+ did not increase as much as the wild-type mice. Indeed, pT286-CaMKII levels did not increase in NMDAR-knockdown mice, whereas, its levels were significantly elevated in wild-type mice [64]. These findings suggested that CaMKII has a key role in nociception in neuropathic pains. (Fig. 1)

Overview of the role of receptor channels and protein kinases in pain sensory neuron hyperactivity. ATP, adenosine triphosphate; CaMK, calcium/calmodulin dependent protein kinase; cAMP, cyclic adenosine monophosphate; DAG. diacylglycerol; IP3, inositol trisphosphate; GPR177, orphan G protein–coupled receptor 177; mTOR, mammalian target of rapamycin; NMDAR, N-methyl-D-aspartate receptor; P, phosphate; P2 × 7R, purinergic 2 × 7 receptor; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol bisphosphate; PIP3, phosphatidylinositol triphosphate; PKA, protein kinase A; PKB, protein kinase B; PKC, protein kinase C; PLC, phospholipase C; SER, smooth endoplasmic reticulum; TRPV1, Transient receptor potential vanilloid 1
Capsaicin cannot be administered orally due to its gastrointestinal side effects. Fajrin et al., investigated 6-shogoad, a chemical agent extracted from ginger with structural similarity to capsaicin, in diabetic rats and found that this agent alleviated thermal allodynia and suppressed the gene expressions of both TRPV1 and NMDAR in the DRG neurons of rats. Interestingly, 6-shogoal treatment intensified insulin immunoreactivity in the pancreatic islets cells of diabetic rats, a unique feature that has not been reported for capsaicin [65].
The central role of NMDAR in pain perception has been shown in rats with induced trigeminal nerve pain as inducing NMDAR enhanced pain perception and inhibition of NMDAR resulted in reduced pain feeling. NMDA acted by stimulating TRPV1 activity as blocking TRPV1 channels abrogated NMDA-evoked pain. Moreover, it was found that NMDA increased the activities of both PKC and CaMKII, and these proteins phosphorylated TRPV1 channels. Interestingly, PKA inhibition resulted in reduced pain perception, however, its mechanism of action was independent from TRPV1 because chemical activation of PKA did not increase phosphorylated TRPV1 levels [66].
Modulating TRPV1 activity also impacts CaMKII, as it has been demonstrated in a rat model of neuropathic pain that silencing TRPV1 suppresses the expression of CaMKII and declines phosphorylated ERK levels in DRG neurons [67]. In addition to CaMKII, the role of CaMKIV in neuropathic pain has also been investigated. Zhao and colleagues reported that CaMKIV inhibition reduced HMGB1 expressions in DRN neurons of diabetic rats, alleviating the thermal hyperalgesia and mechanical allodynia. Therefore, dual blockade of CaMKII and CaMKIV can produce more efficient analgesic effects [68].
Conclusion
Diabetic neuropathic pain is an important sequela of diabetic neuropathy which deeply affects patient’s life quality. It is related to abnormally increased activity of pain sensory neurons and therefore neither steroidal nor non-steroidal anti-inflammatory drugs are effective in alleviating DNP. Therapeutic goal is to specifically suppress pain sensory neurons, without affecting motor as well as other sensory neurons. Opioid analgesics are not routinely prescribed for DNP due to their broad spectrum of side effects. Currently, DNP is mainly managed by duloxetine or venlafaxine (serotonin–norepinephrine reuptake inhibitor antidepressants) and pregabalin, or gabapentin (gabapentinoid anticonvulsants). Again, these agents are non-specific and are switched if there is no response or if side effects develop. TRPV1, P2 × 7R, and NMDAR are receptor channels mainly expressed on pain sensory neurons and their modulation attenuates diabetes associated pain with no effect on other nervous functions; for instance, the safety and efficacy of %8 capsaicin dermal patch has been proved in large scale clinical studies. However, the majority of studies are at the experimental levels and conducting clinical trials to evaluate the safety and efficacy of these therapeutic modalities are highly warranted.
Data Availability
This article does not contain any studies with human participants or animals performed by any of the authors.
References
Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic neuropathy. Nat reviews Disease primers. 2019;5:1–18.
Elafros MA, Andersen H, Bennett DL, Savelieff MG, Viswanathan V, Callaghan BC, et al. Towards prevention of diabetic peripheral neuropathy: clinical presentation, pathogenesis, and new treatments. Lancet Neurol. 2022;21:922–36.
Gylfadottir SS, Itani M, Kristensen AG, Karlsson P, Krøigård T, Bennett DL, et al. The characteristics of pain and dysesthesia in patients with diabetic polyneuropathy. PLoS ONE. 2022;17:e0263831.
Tesfaye S, Boulton AJ, Dickenson AH. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care. 2013;36:2456–65.
Davies M, Brophy S, Williams R, Taylor A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care. 2006;29:1518–22.
Feldman EL, Nave K-A, Jensen TS, Bennett DL. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron. 2017;93:1296–313.
Gupta M, Knezevic NN, Abd-Elsayed A, Ray M, Patel K, Chowdhury B. Treatment of painful diabetic neuropathy—a narrative review of pharmacological and interventional approaches. Biomedicines. 2021;9:573.
Graham RD, Bruns TM, Duan B, Lempka SF. Dorsal root ganglion stimulation for chronic pain modulates Aβ-fiber activity but not C-fiber activity: a computational modeling study. Clin Neurophysiol. 2019;130:941–51.
Roskoski R Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1–23.
Ardito F, Giuliani M, Perrone D, Troiano G, Lo Muzio L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy. Int J Mol Med. 2017;40:271–80.
Paoletti P, Ellis-Davies GC, Mourot A. Optical control of neuronal ion channels and receptors. Nat Rev Neurosci. 2019;20:514–32.
Woll KA, Van Petegem F. Calcium-release channels: structure and function of IP3 receptors and ryanodine receptors. Physiol Rev. 2022;102:209–68.
Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol. 2010;72:95–121.
Fallah HP, Ahuja E, Lin H, Qi J, He Q, Gao S, et al. A review on the role of TRP channels and their potential as drug Targets_An Insight into the TRP Channel Drug Discovery Methodologies. Front Pharmacol. 2022;13:1784.
Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–10.
Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–7.
Kamei J, Zushida K, Morita K, Sasaki M, Tanaka S-i. Role of vanilloid VR1 receptor in thermal allodynia and hyperalgesia in diabetic mice. Eur J Pharmacol. 2001;422:83–6.
Forst T, Pohlmann T, Kunt T, Goitom K, Schulz G, Löbig M, et al. The influence of local capsaicin treatment on small nerve fibre function and neurovascular control in symptomatic diabetic neuropathy. Acta Diabetol. 2002;39:1–6.
Vinik AI, Perrot S, Vinik EJ, Pazdera L, Jacobs H, Stoker M, et al. Capsaicin 8% patch repeat treatment plus standard of care (SOC) versus SOC alone in painful diabetic peripheral neuropathy: a randomised, 52-week, open-label, safety study. BMC Neurol. 2016;16:1–14.
Simpson DM, Robinson-Papp J, Van J, Stoker M, Jacobs H, Snijder RJ, et al. Capsaicin 8% patch in painful diabetic peripheral neuropathy: a randomized, double-blind, placebo-controlled study. J Pain. 2017;18:42–53.
Walker KM, Urban L, Medhurst SJ, Patel S, Panesar M, Fox AJ, et al. The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;304:56–62.
Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–29.
Reid G, Flonta M-L. Ion channels activated by cold and menthol in cultured rat dorsal root ganglion neurones. Neurosci Lett. 2002;324:164–8.
Liu B, Qin F. Functional control of cold-and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4, 5-bisphosphate. J Neurosci. 2005;25:1674–81.
Pabbidi MR, Premkumar LS. Role of transient receptor potential channels Trpv1 and Trpm8 in diabetic peripheral neuropathy. Journal of diabetes and treatment 2017; 2017.
Duan B, Wu L-J, Yu Y-Q, Ding Y, Jing L, Xu L, et al. Upregulation of acid-sensing ion channel ASIC1a in spinal dorsal horn neurons contributes to inflammatory pain hypersensitivity. J Neurosci. 2007;27:11139–48.
Bektur E, Şahin E, Ceyhan E, Donmez DB, Canbek M, Baycu C, et al. Beneficial effect of mirtazapine on diabetes-induced hyperalgesia: involvement of TRPV1 and ASIC1 channels in the spinal cord and dorsal root ganglion. Neurol Res. 2019;41:544–53.
Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015;18:145–53.
Xie Y-K, Luo H, Zhang S-X, Chen X-Y, Guo R, Qiu X-Y, et al. GPR177 in A-fiber sensory neurons drives diabetic neuropathic pain via WNT-mediated TRPV1 activation. Sci Transl Med. 2022;14:eabh2557.
Wei L, Caseley E, Li D, Jiang L-H. ATP-induced P2X receptor-dependent large pore formation: how much do we know? Frontiers Media SA; 2016. p. 5.
Janks L, Sharma CV, Egan TM. A central role for P2X7 receptors in human microglia. J Neuroinflamm. 2018;15:1–18.
Wang A, Shi X, Yu R, Qiao B, Yang R, Xu C. The P2X7 Receptor Is Involved in Diabetic Neuropathic Pain Hypersensitivity Mediated by TRPV1 in the Rat Dorsal Root Ganglion.Frontiers in Molecular Neuroscience2021:104.
Chen L, Wang H, Xing J, Shi X, Huang H, Huang J, et al. Silencing P2X7R alleviates Diabetic Neuropathic Pain Involving TRPV1 via PKCε/P38MAPK/NF-κB signaling pathway in rats. Int J Mol Sci. 2022;23:14141.
Wilkerson JL, Alberti LB, Kerwin AA, Ledent CA, Thakur GA, Makriyannis A, et al. Peripheral versus central mechanisms of the cannabinoid type 2 receptor agonist AM1710 in a mouse model of neuropathic pain. Brain and behavior. 2020;10:e01850.
Kao D-J, Li AH, Chen J-C, Luo R-S, Chen Y-L, Lu J-C, et al. CC chemokine ligand 2 upregulates the current density and expression of TRPV1 channels and Nav1. 8 sodium channels in dorsal root ganglion neurons. J Neuroinflamm. 2012;9:1–13.
Wilkerson JL, Alberti LB, Thakur GA, Makriyannis A, Milligan ED. Peripherally administered cannabinoid receptor 2 (CB2R) agonists lose anti-allodynic effects in TRPV1 knockout mice, while intrathecal administration leads to anti-allodynia and reduced GFAP, CCL2 and TRPV1 expression in the dorsal spinal cord and DRG. Brain Res. 2022;1774:147721.
Newton AC. Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol. 2018;53:208–30.
Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circul Res. 2010;106:1319–31.
Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–90.
Hong S, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J Biol Chem. 2005;280:618–27.
Pabbidi RM, Yu S-Q, Peng S, Khardori R, Pauza ME, Premkumar LS. Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol Pain. 2008;4:1–17.
Ramírez-Coronel AA, Abdu WJ, Alshahrani SH, Treve M, Jalil AT, Alkhayyat AS, Singer N. Childhood obesity risk increases with increased screen time: a systematic review and dose–response meta-analysis. J Health Popul Nutr. 2023;42(1):5.
Lam D, Momeni Z, Theaker M, Jagadeeshan S, Yamamoto Y, Ianowski JP, et al. RAGE-dependent potentiation of TRPV1 currents in sensory neurons exposed to high glucose. PLoS ONE. 2018;13:e0193312.
Johnson K, Doucette A, Edwards A, Watts VJ, Klein AH. Reduced activity of Adenylyl Cyclase 1 Attenuates Morphine Induced Hyperalgesia and Inflammatory Pain in Mice. 2020.
Herrmann S, Rajab H, Christ I, Schirdewahn C, Höfler D, Fischer MJ, et al. Protein kinase a regulates inflammatory pain sensitization by modulating HCN2 channel activity in nociceptive sensory neurons. Pain. 2017;158:2012–24.
Kareem AA, Sabhan AH. The significant relationship between a prostatitis and a high Level of glycated hemoglobin in non-Insulin dependent Diabetes Mellitus in Al-Diwaniah Province. J. Biomed. Biochem. 2022:1(2):25–28. https://doi.org/10.57238/jbb.2022.5455.1007
Xu X, Ren Z, Tang Y, Zilundu PL, Zhou Y, Li W et al. Dexmedetomidine attenuates hyperalgesia induced by brachial plexus root avulsion by restoring the GLT-1 function via PKA signaling. 2022.
Bhave G, Zhu W, Wang H, Brasier D, Oxford GS, Gereau IVRW. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–31.
Gunn RM, Hailes HC. Insights into the PI3-K-PKB-mTOR signalling pathway from small molecules. J Chem Biol. 2008;1:49–62.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405.
Guo J-R, Wang H, Jin X-J, Jia D-L, Zhou X, Tao Q. Effect and mechanism of inhibition of PI3K/Akt/mTOR signal pathway on chronic neuropathic pain and spinal microglia in a rat model of chronic constriction injury. Oncotarget. 2017;8:52923.
Chen H, Hu Y, Xie K, Chen Y, Wang H, Bian Y, et al. Effect of autophagy on allodynia, hyperalgesia and astrocyte activation in a rat model of neuropathic pain. Int J Mol Med. 2018;42:2009–19.
Liu K, Yang Y, Zhou F, Xiao Y, Shi L. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy and relieves hyperalgesia in diabetic rats. NeuroReport. 2020;31:644–9.
Bayer KU, Schulman H. CaM kinase: still inspiring at 40. Neuron. 2019;103:380–94.
Qian Y, Xia T, Cui Y, Chu S, Ma Z, Gu X. The role of CaMKII in neuropathic pain and fear memory in chronic constriction injury in rats. Int J Neurosci. 2019;129:148–56.
Yu H, Pan B, Weyer A, Wu H-E, Meng J, Fischer G, et al. CaMKII controls whether touch is painful. J Neurosci. 2015;35:14086–102.
Morris EP, Török K. Oligomeric structure of α-calmodulin-dependent protein kinase II. J Mol Biol. 2001;308:1–8.
Boric M, Kadic AJ, Puljak L. The expression of calcium/calmodulin-dependent protein kinase II in the dorsal horns of rats with type 1 and type 2 diabetes. Neurosci Lett. 2014;579:151–6.
Ferhatovic L, Banozic A, Kostic S, Kurir TT, Novak A, Vrdoljak L, et al. Expression of calcium/calmodulin-dependent protein kinase II and pain-related behavior in rat models of type 1 and type 2 diabetes. Anesth Analgesia. 2013;116:712–21.
Jerić M, Vuica A, Borić M, Puljak L, Kadić AJ, Grković I, et al. Diabetes mellitus affects activity of calcium/calmodulin-dependent protein kinase II alpha in rat trigeminal ganglia. J Chem Neuroanat. 2015;64:12–9.
Bian H, Yu L-C. Intra-nucleus accumbens administration of the calcium/calmodulin-dependent protein kinase II inhibitor AIP induced antinociception in rats with mononeuropathy. Neurosci Lett. 2015;599:129–32.
He X-f, Kang Y-r, Fei X-y, Chen L-h, Li X, Ma Y-q et al. Inhibition of phosphorylated calcium/calmodulin-dependent protein kinase IIα relieves streptozotocin-induced diabetic neuropathic pain through regulation of P2X3 receptor in dorsal root ganglia.Purinergic Signalling2022:1–13.
Cairns BE, Svensson P, Wang K, Hupfeld S, Graven-Nielsen T, Sessle BJ, et al. Activation of peripheral NMDA receptors contributes to human pain and rat afferent discharges evoked by injection of glutamate into the masseter muscle. J Neurophysiol. 2003;90:2098–105.
Matsumura S, Kunori S, Mabuchi T, Katano T, Nakazawa T, Abe T, et al. Impairment of CaMKII activation and attenuation of neuropathic pain in mice lacking NR2B phosphorylated at Tyr1472. Eur J Neurosci. 2010;32:798–810.
Omear HA. Novel SNPs of TNF-a and IL-6 that Regulate Serum Level in Obese Patients. J. Biomed. Biochem. 2023:2(1):7–20. https://doi.org/10.57238/jbb.2023.6398.1025
Lee J, Saloman JL, Weiland G, Auh Q-S, Chung M-K, Ro JY. Functional interactions between NMDA receptors and TRPV1 in trigeminal sensory neurons mediate mechanical hyperalgesia in the rat masseter muscle. Pain. 2012;153:1514–24.
Guo S-H, Lin J-P, Huang L-E, Yang Y, Chen C-Q, Li N-N, et al. Silencing of spinal Trpv1 attenuates neuropathic pain in rats by inhibiting CAMKII expression and ERK2 phosphorylation. Sci Rep. 2019;9:1–9.
Zhao X, Shen L, Xu L, Wang Z, Ma C, Huang Y. Inhibition of CaMKIV relieves streptozotocin-induced diabetic neuropathic pain through regulation of HMGB1. BMC Anesthesiol. 2015;16:1–8.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Mustafa Gheni Taher and Mazin Razooqi Mohammed gave the concept. Muthanna Abdulkhader Salh Al-Mahdawi and Noor Kareem Assi Halaf wrote the manuscript. Abduladheem Turki Jalil prepared the figure. Tahani Alsandook scientifically revised the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to publish
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Taher, M.G., Mohammed, M.R., Al-Mahdawi, M.A.S. et al. The role of protein kinases in diabetic neuropathic pain: an update review. J Diabetes Metab Disord 22, 147–154 (2023). https://doi.org/10.1007/s40200-023-01217-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40200-023-01217-1