
Abstract
Hypoxic–ischemic encephalopathy, also referred as HIE, is a type of brain injury or damage that is caused by a lack of oxygen to the brain during neonatal period. The incidence is approximately 1.5 cases per 1000 live births in developed countries. In low and middle-income countries, the incidence is much higher (10‒20 per 1000 live births). The treatment for neonatal HIE is hypothermia that is only partially effective (not more than 50% of the neonates treated achieve an improved outcome). HIE pathophysiology involves oxidative stress, mitochondrial energy production failure, glutaminergic excitotoxicity, and apoptosis. So, in the last years, many studies have focused on peptides that act somewhere in the pathway activated by severe anoxic injury leading to HIE. This review describes the pathophysiology of perinatal HIE and the mechanisms that could be the target of innovative HIE treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypoxic–ischemic encephalopathy (HIE) is a term used to describe the complex physiological, cellular, and molecular changes resulting from a severe anoxic brain injury during neonatal period. This can lead to premature mortality or a variety of life-long morbidities, including acute symptoms such as seizures, alteration of consciousness, weak breathing, poor muscle tone or metabolic derangement as well as chronic conditions such as cerebral palsy, epilepsy, intellectual disability, and behavioral disorders [1, 2].
HIE has an incidence of approximately 1.5 cases per 1000 live births in developed countries and 10‒20 per 1000 live births in low and middle-income countries [3,4,5].
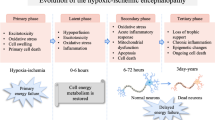
HIE is not a single event, rather an ongoing process causing neuronal cell deaths over hours to day after the initial injury. In fact, it is possible to recognize three distinct phases [6]. The first immediate phase (primary neuronal death) is determined by the primary energy failure during hypoxic–ischemic (HI) event, resulting in oxidative metabolism failure, cytotoxic edema, and accumulation of excitotoxins [7]. After restoration of cerebral circulation, the second phase begins (a latent phase lasting about 6 h). Between 6 and 15 h after the HI insult, occurs the secondary energy failure (delayed neuronal death) that is associated with encephalopathy and increased seizure activity. The mechanisms involved in this phase include excitotoxicity, apoptosis, and microglial activation [8].
Therapeutic hypothermia is the current standard of care for infants with HIE, cooling the whole body to a core temperature of 33.5 °C for 72 h starting within 6 h of birth [9]. According to scientific evidences, therapeutic hypothermia inhibits key steps in the excito-oxidative cascade [1]. However, it does not provide complete neuroprotection and it is only partially effective. Even after treatment, there is a high prevalence of neurologic morbidity and mortality, affecting 40–50% of neonates who underwent therapeutic hypothermia [10].
In the last years, many studies have focused on research of new treatments for HIE that, together with hypothermia, could enhance neuroprotection and lower adverse outcomes [11]. Since HIE pathophysiology involves oxidative stress, mitochondrial energy production failure, glutaminergic excitotoxicity, and apoptosis [12], these researches are focusing on peptides that act somewhere in the pathway activated by severe anoxic injury leading to HIE.
In this review, we conducted a literature search to better clarify the pathophysiology of perinatal HIE and the inflammatory mechanisms involved in it, those mechanisms that could be the target of innovative HIE treatments.
Materials and methods
A review of the literature was conducted to identify the most relevant studies reported in the English since 2017 on PubMed MEDLINE electronic database. Based on the abstracts, we made a selection of these studies, focusing on articles that concerned HIE pathophysiology and inflammatory mechanisms. The exclusion criteria were articles not in English and not relevant to the review and abstracts (Fig. 1).

The screen procedure for article inclusion for review
The keywords used were “hypoxic-ischemic encephalopathy”, “Hypoxic-Ischemic Injury”, “developing brain”, “neonatal brain”, “neonatal”, “perinatal”, “pathophysiology”. Different combinations of the terms were used. Moreover, references in each article were searched to identify potentially missed studies.
Results and discussion
Pathophysiology
Fetal brain requires a constant supply of energy in form of ATP that obtains metabolizing lactate, ketone bodies, and glucose. Comparing to adult, fetal brain has a greater ability to tolerate hypoxia–ischemia (HI), due to its capacity to reserve energy when needed. However, in case of a critical depletion of ATP also, the fetal brain becomes susceptible to injury. This critical ATP depletion can be caused by prolonged or sudden and important HI, originating from various conditions (i.e. chronic maternal hypoxia, pre-eclampsia, umbilical cord knotting, umbilical cord prolapse, shoulder dystocia and placental abruption) leading to an impairment of oxygenated cerebral blood flow to the fetus causing systemic and cellular responses [13]. HI brain injury is an ongoing process composed by several different phases. When the primary critical energy failure occurs, an uncontrolled release of excitatory neurotransmitters begins, starting the ischemic cascade that damages neuronal cells (both at cytoplasmic and mitochondrial level), disrupts the brain–blood barrier (the amount of membrane peroxidation is directly related to the severity of ATP depletion) and activates an important inflammatory response. These result in failure of oxidative metabolism, cytotoxic edema, and accumulation of excitotoxins [5]. After the cerebral circulation restoration, a latent phase begins, lasting around 6 h, followed by (6–15 h after HI) a secondary energy failure that can last for days. Typical of this phase is seizures, renewed cytotoxic edema, release of excitotoxins, impaired cerebral oxidative energy metabolism, and finally, neuronal cell death [14].
HIE pathophysiology (Fig. 2) can be outlined in five main events, linked one to the others: oxidative stress, intracellular Ca2+ accumulation, mitochondrial dysfunction, excitotoxicity, and inflammation [13].

Pathophysiology of HIE
Oxidative stress
Fetal brain is an environment particularly susceptible to oxidative stress, since unsaturated fatty acids and metals catalyzing free radical reactions are heavily present, while antioxidant levels are low [15]. Oxidative stress and reactive oxygen species (ROS) produced by HI cause an important damage to lipids, proteins, and nucleic acids, leading to lipid and protein oxidation and DNA degeneration.
Normally ROS, mainly generated in mitochondria, are cleared by superoxide dismutase and glutathione peroxidase. During HI, ROS cannot be immediately eliminated by antioxidant enzymes because of interrupted metabolism, thus an excessive ROS accumulation occurs [16]. This, together with oxidative modification of lipids, proteins, and DNA leads to metabolic failure and mitochondrial dysfunction following newborn hypoxic–ischemic insult.
In HIE, ROS are generated from mitochondrial electron transport chain, NADPH oxidases, xanthine oxidase, arachidonic acid (12/15 lipoxygenase), and nitric oxide (NO) synthase.
NO synthase is activated by a mechanism enabled by hypoxia-induced energy depletion. This causes alterations of the NMDA receptor ion channel, inducing influx of Ca2+ into the cytoplasm. Increase of intracellular Ca2+ leads to the activation of NO synthase via a Ca2+/calmodulin-dependent mechanism, generating NO free radicals.
NO produced during hypoxia has several adverse neuronal effect through different mechanisms. First, NO increases the expression of apoptotic proteins, activating CREB protein, through a NO-mediated pathway involving CaM kinase IV activity and CaM kinase IV-dependent phosphorylation of CREB protein, thus leading to increased expression of proapoptotic genes [15, 17]. Second, NO reaction with superoxide generates peroxynitrite, a toxic free radical known to alter cell membranes, to interfere with protein and receptor activity and to activate pre-apoptotic pathway [18]. In addition, NO causes lipid peroxidation, protein oxidation and nitration of nuclear membranes, DNA damage and increasing in intranuclear Ca2+ [15].
Because of all these adverse neuronal effects, the inhibition of NO synthase has been a target in HIE studies on neuroprotection [6, 15, 19].
Moreover, a surplus of ROS limits glucose metabolism. In fact, excessive ROS accumulation inhibits pyruvate dehydrogenase complex (PDHC) activity. Because of this alteration in PDHC activity and the impairment of reducing power transportation from glycolysis-generated NADH to mitochondria, lactate dehydrogenase, normally activated by elevated Ca2+, will use NADH to form massive amounts of lactate in cytosol. Acidosis is the obvious consequence of this lactate accumulation in neurons during HI [20].
Intracellular Ca2+ accumulation
Ca2+ extracellular concentration can reach up to 1–2 Mm, while intracellular Ca2+ is normally very low (about 100 nM) [21]. Several mechanisms contributes in maintaining the intracellular Ca2+ concentration (i.e. Ca2+ release from endoplasmic reticulum; Ca2+ release through Ca2+–ATPase or Na+–Ca2+ exchanger in cell membrane; Ca2+ transportation into mitochondria through an electrophoretic uniporter, entry of extracellular Ca2+ into cytosol through NMDA receptors or voltage-gated Ca2+ channels). NMDA receptor channel is probably one of the main pathway in intracellular Ca2+ accumulations, as shown by studies on NMDA antagonists, found to block Ca2+ entry in neurons and thus significantly attenuating neurodegeneration after HI [22].
In HI condition, the deprivation of oxygen and glucose caused by abnormal blood circulation triggers the overstimulation of glutamate release. Glutamate stimulates the opening of NMDA receptor channels, allowing Ca2+ to flow inside neurons [20] (Fig. 3).

Effect of Ca2+ surge in neurons
High intracellular Ca2+ concentration has various effects: as seen before, activates NO synthase via a Ca2+/calmodulin-dependent mechanism; moreover, it can induce mitochondrial dysfunction and mediate irreversible immature neuron death by the activation of several Ca2+-dependent proteins. Among these, there are proteins, as calpains, involved in cell remodeling, membrane destruction and neuron degeneration [23, 24]. Furthermore, increase in intramitocondrial Ca2+ concentration, together with ROS, contributes to mitochondrial permeability transition and, consequently, to nicotinamide adenine dinucleotide loss, important for ROS detoxification and cellular energy metabolic processes [20].
Mitochondrial dysfunction
Mitochondria carry out many important functions and their alteration assumes a central role in HI-induced neurodegenerations, thus determining the destiny of cells subjected to HI. Mitochondria produce energy indispensable for multiple cellular activities, a process that generates ROS, and are also the most important intracellular Ca2+ buffers. Their dysfunction, indeed, can lead to mitochondrial energy failure and, consequently, to intracellular Ca2+ accumulation. This is caused by the reduction of the energy needed to maintain membrane ion gradient and worsened by the opening, in HI condition, of NMDA receptors and voltage-gated Ca2+ channels [25, 26]. Moreover, the normal functioning of electron transport chain (ETC) is interrupted because of ROS excess present in HI, thus amplifying the production of mitochondria-free species [27]. Oxidative modification of proteins and lipids of mitochondrial inner membrane alters its permeability since membrane depolarization occurs. This alteration uncouples oxidative phosphorylation processes resulting in ATP production deficiency. This drastic energetic failure contributes to cell membrane depolarization and, consequently, to Ca2+ influx, instituting a sort of vicious cycle [20].
Therefore, mitochondrial dysfunction can cause a series of lethal problems, as oxidative stress, intracellular Ca2+ accumulation, and mitochondrial energy failure that lead to neuron apoptosis and, consequently, neurodegeneration.
Studies based on mitochondrial energy failure have focused on AMPK role [28]. AMPK is a sort of cell energy sensor and is activated when there is an imbalance in the AMP:ATP ratio, as happens in HI. Once activated, AMPK acts to inhibit energy consumption (fatty acid/cholesterol synthesis) and promote energy production (e.g. glycolysis), trying to restore energy balance. AMPK activation is due to two kinases: LKB1 and CaMKKβ [29]. The latter is activated by intracellular Ca2+ excess. Moreover, another AMPK mediates apoptosis during excitotoxicity, through the expression of the proapoptotic protein Bim [30]. More specifically, excitotoxicity and ROS overload seen in HI cause calcium surge that not only activates CaMKKβ and, consequently, AMPK, but also, at the same time, challenges the mitochondrial respiratory chain. In this way, the imbalance in AMP:ATP ratio increases and AMPK activation is reinforced. This AMPK activation restores energy balance, explaining the return to basal level of ATP after the primary energy failure [28]. Later, the other mechanisms involved in HI, such as inflammation, damage mitochondrial function once again, causing the second energy failure and the rebound of AMPK over activation. These deleterious events could cause a final mitochondrial challenge, leading to mitochondria membrane permeabilization and cellular apoptosis.
Apoptosis too, as many other HI events, is linked to Ca2+ accumulation in cytoplasm together with ROS. As a matter of fact, accumulation of Ca2+ and ROS activates the mitochondrial permeability transition pore (MPTP) within the mitochondria inner membrane, inducing mitochondrial permeability transition (MPT). The role of MPTP in HI has been confirmed by studies on MPTP inhibitor cyclosporine A. It helps in protecting against HI neurodegeneration by binding a component of MPTP (cyclophilin D, CypD) thus interrupting its involvement in MPTP construction [31, 32]. Despite these studies, the exact role and entity of MPT involved in neonatal HIE is still unclear and the CypD-based treatment remains controversial.
MPTP releases in cytoplasm nicotinamide adenine dinucleotide (NAD+), a cofactor important for energy metabolic reactions and ROS detoxification. Mitochondrial distress will determine the formation of Bax/Bak megapores inside the mitochondria outer membrane, causing release of mitochondrial contents, including proapoptotic proteins such as cytochrome c (CytC), apoptosis-inducing factor (AIF), endonuclease (endo) G and Smac/Diablo. Each protein has different downstream targets, but they all contribute to cell death. CytC and Smac/Diablo triggers caspase activation, determining an irreversible apoptosis [33], while AIF, after interacting with cyclophilin A, translocates to the nucleus where it causes DNA fragmentation [34]. Caspases, ROS, and AIF induce DNA fragmentation that activates poly (ADP-ribose) polymerase (PARP), a DNA repairing enzyme. The DNA repairing process consumes NAD+ . This NAD+ loss plus oxidative failure will worsen mitochondrial dysfunction, by depleting NAD+ needed to maintain mitochondrial energetics, and will extend the impairment of neonatal hypoxic–ischemic brain injury.
During neurodegeneration, two forms of neuron death could happen: necrosis or apoptosis.
Many studies, focusing on high expression of proapoptotic proteins, such as Caspase-3, Bax, and BCl, have elucidated that apoptosis is the more prevalent form in immature brain injury compared with adult models [20, 35].
Subsequently, largely due to advances in cell biology and to experimental animal studies, emphasis has been switched also to autophagy mediated by programmed cell death (PCD) mechanisms as important forms of degeneration in HIE. For instance, preclinical evidence has now shown that hypoxic–ischaemia exacerbates autophagic flux in neonatal rats and leads to learning and memory impairment. Moreover, recent clinical data showed an upregulation of MALAT1 after HIE which controls the HIF-1α axis and autophagic cell death [36,37,38,39,40].
Accordingly, several recent studies have focused on mitochondrial-related therapeutic targets, such as Bax-inhibiting peptide (BIP), PARP inhibition, MPT inhibition, NAD administration, with promising results for future applications [20]. In summary, the mitochondrial dysfunction is the key point of neurodegeneration in HIE.
Excitotoxicity
Excitotoxicity, a term first used in the 1970s, refers to cell death mediated by excessive stimulation of extracellular excitatory amino acid receptors [41]. These receptors mediate the glutamate physiologic excitatory effects. During HI, they are excessively stimulated by elevated glutamate levels and membrane depolarization, leading to their inappropriate opening and, consequently, to a lethal flow of Ca2+ inside neurons. In fact, following prolonged HI, cellular homeostasis is disrupted due to ATP depletion and to the incapacity to maintain ionic gradients. This causes neuronal depolarization and glutamate release into the synaptic cleft, with an overload of extracellular glutamate, due also to a reduced activity of the glial pumps that normally keep synaptic glutamate levels low [26]. This leads to excitotoxicity in neurons and other cells (glial progenitor cells) that express glutamate receptors [13], with their inappropriate opening. As depicted above, the consequent Ca2+ flow inside neurons and glial progenitor cells results in activation of calcium-dependent proteases, lipases, and deoxyribonucleases, ROS production, oxidative stress, cytotoxic edema, mitochondrial dysfunction and, eventually, stimulation of proapoptotic cellular pathways.
Inflammation
In the immature brain, within minutes after an HI injury, an innate immune response takes place. Microglia, neutrophils, lymphocytes, cytokines, selections, and immunoglobulins, all have a role in the inflammatory process activated by hypoxia.
Microglia
Microglia provides immuno-surveillance to the brain. When HI happens, microglia activates and develops macrophage-like abilities, such as phagocytosis, antigen presentation, inflammatory and anti-inflammatory cytokines production and matrix metalloproteinases (MMPs) release, which lead to blood–brain barrier (BBB) breakdown [42]. Consequently, peripheral leukocytes have free access to the brain, and the normally immune-privileged brain environment is exposed to systemic responses, further exacerbating inflammation and brain damages [8].
The role of microglia in HIE is supported by different studies: some, focusing on postmortem examinations of neonatal brains, have found that patient died from HIE had a dense infiltrate of microglia in the hippocampal dentate gyrus, whereas those neonates who died of other acute causes (trauma or sepsis) had significantly fewer microglia [43]. Other studies, instead, have focused on cytokines released by activated microglia after HI, e.g. IL-18 and Caspase-1. Genetic deletion of IL-18 or of Caspase-1 attenuates brain injury [44]. However, it is important to highlight that no intervention targets microglia selectively. Indeed, pharmacological depletion of microglial prior to neonatal stroke aggravates rather than improves outcome, exacerbating the release of inflammatory cytokines. This suggests that at least a part of microglia has beneficial effects [45]. Indeed, for example, microglial phagocytosis of remains has been hypothesyzed to be critical for tissue recovery during the second delayed phase after HI. These contrasting findings can be explained with different microglial activation phenotypes. Some microglia participates in acute early pro-inflammatory responses and aggravates injury, whereas others might be involved in the late anti-inflammatory responses and protect against injury [44]. According to literature [8, 46], indeed, there are two microglial activation phenotypes. Classical activation (M1), responsible of innate immune response, that leads to the production of cytokines, chemokines, and reactive intermediates. Another activation phenotypes (M2) responsible of anti-inflammatory signaling (M2a) and clearance of ROS and nitrogen species (M2b). The M1 phenotypes can lead to increases neuronal death compared to M2, this is why nowadays there is a growing interest in controlling the classical activation pattern of microglia.
Astrocytes
As well as microglia, astrocytes are activated within minutes after injury by pro-inflammatory mediators, cytokines, and ROS secreted by injured cells. Astrocytes have opposite role in HI: from one side, they release glutathione and superoxide dismutase (SOD) [47] and enhanced extra-synaptic glutamate uptake [48]; from the other, they produce and release pro-inflammatory cytokines (IL-6, TNF-α, IL-1α, and β and interferon γ). This cytokines exacerbate HI injury directly inducing neuronal cells apoptosis, inhibiting neurogenesis, and increasing toxic NO levels [8]. Moreover, astrocytes release chemokines that attracts immune cells to the ischemic site, thus worsening the brain injury.
Neutrophils
Neutrophils can worsen brain injury determined by HI through multiple mechanisms, such as ROS production, release of cytotoxic agents, MMP-9 secretion, and decreased microvascular flow due to neutrophils accumulation inside vessels. Differently from adult brain, where neutrophil infiltration can be massive, neonates have a lower capacity to mount a neutrophil response after ischemia, as proven by the fact that neonatal neutrophils have reduced extravasation from blood vessels [8].
This finding can explain why treatment based on neutrophil inhibitory factor, neuroprotective in adult animals, is not so efficacious in neonatal rats [49].
Lymphocytes and mast cells
Differently from microglia and neutrophils, the role of lymphocytes and mast cells in HI is not yet totally clear. Lymphocyte infiltration of neonatal brain after HI seems to be, differently from what happens in adult, less profound or only briefly present, reflecting the immaturity of lymphoid progenitor cells. Studies suggest that a lymphocytic response is involved in the more chronic immunoinflammatory activation following HIE, since CD4 have been found in damaged areas 7 days after HIE, persisting there for 14–35 days [50]. However, it is still unknown whether this lymphocytic presence enhances injury or, conversely, neuron restoration.
Mast cells, on the other side, seem to promote inflammation acutely after damage and to contributes to excitotoxic injury early releasing TNF and exacerbating transforming growth factor β1 (TGFβ1) toxicity. However, the contributions of mast cells to the ongoing evolution of damage or reparation are still unclear.
Cytokines and chemokines
Activated microglia, astrocytes, and endothelial cells release chemokines and pro-inflammatory cytokines, such as tumor necrosis factor α (TNF-α), transforming growth factorβ (TGF-β), IL-1, IL-6, IL-8, and IL-10. Cytokines attract leukocytes and facilitate their stop in the brain stimulating the production of adhesion molecules on leukocytes and endothelial cells. TNF-α and IL-1 β, often expressed simultaneously, are two of the best-characterized early response cytokines and are secreted by microglia, astrocytes, and neurons [8, 51]. These cytokines determine the accumulation of inflammatory cells in the injured brain attracting them. Neonates with HIE have higher level of TNF-α and IL-1 β in peripheral blood samples compared to control and IL-1 β correlates positively with HIE severity [52]. Studies on administration of IL-1 receptor antagonist in HIE models have demonstrated the neuroprotective potential of this treatment, reaffirming the damaging involvement of this cytokine in HIE [53, 54].
Chemokines also play a central role in cerebral injury in HIE controlling inflammatory cell traffic. They act through specific receptors belonging to the family of G-protein-coupled receptors [55]. Studies on HIE in rats have demonstrated the upregulation of α- and β-chemokines before the expression of lymphocytes receptor in the damaged area [50].
Neuroprotective agents for neonatal hypoxic–ischemic brain injury and future prospects with neural regeneration agents
Different potential neuroprotective treatments are being studied to prevent the cascade of injurious effects after hypoxia–ischemia. These promising neuroprotective agents tested on animal models and pilot clinical studies of neonatal H–I brain injury target different phases of injury: the early phase of excitotoxicity, oxidative stress and apoptosis as well as late-phase inflammatory reaction and neuronal and oligodendrocyte regeneration.
The present neuroprotective agents for early treatment of neonatal hypoxic–ischemic brain injury are classified into four categories according to currently known mechanisms: antiexcitotoxicity, antioxidation, anti-inflammation, and antiapoptosis [56, 57].
Xenon
Xenon has two therapeutic effects (antiexcitotoxic and antiapoptotic). Acute hypoxic–ischemic insult leads to NMDA receptor activation. Xenon inhibits NMDA signaling and thus may play a role in reducing the acute cell injury. Studies of birth asphyxia in the piglet model suggest a benefit in the association of treatment with hypothermia and xenon [58, 59]. But a proof of concept, open-label, randomised controlled trial (Total Body hypothermia plus Xenon-TOBY-Xe) [60] concluded that xenon is unlikely to enhance the neuroprotective effect of cooling after birth asphyxia. Additionally, considering that xenon is an expensive noble gas (it requires a specialized delivery system) [61], alternative therapies are currently being evaluated.
Argon
Argon is a less expensive noble gas with antiapoptotic effect that has demonstrated significant neuroprotection in animal models of HIE. An extensive piglet study of perinatal asphyxia showed that inhaled 45–50% argon augments hypothermic brain protection [62].
Melatonin and erythropoietin (EPO)
Melatonin and erythropoietin (EPO) have three therapeutic effects (anti-inflammatory, antioxidative, and antiapoptotic). In a randomized controlled pilot trial evaluating melatonin with cooling in term infants with HIE, compared to controls, combination of melatonin to therapeutic hypothermia was efficacious in reducing oxidative stress and improving survival with favorable neurodevelopmental outcomes at 6 months of age [63]. Melatonin seems to be safe and beneficial in protecting neonatal brains from perinatal HIE. However, larger randomized controlled trials in humans are required.
Erythropoietin and darbepoetin (a long acting erythropoietin analogue that offers the additional benefit of once weekly administration) have neuroprotective properties in animal models of hypoxic–ischemic brain injury and neonatal stroke [57]. High doses of erythropoietin, both in conjunction with hypothermia and as monotherapy, have shown promise in preliminary randomized trials for reducing brain injury and improving motor outcomes in infants with HIE [64]. A small randomized trial evaluating the use of darbepoietin as adjunctive therapy to hypothermia in the first 12 h of life and repeated at 1 week of life demonstrated a good safety profile of this medication [65]. However, confirmation from larger trials is needed.
Allopurinol
Allopurinol is a xanthine oxidase inhibitor with one therapeutic effect (antioxidative). It reduces the production of oxygen radicals as superoxide, which contributes to secondary energy failure and apoptosis in neurons and glial cells after reperfusion of hypoxic brain tissue.
Preclinical studies in animal models of HIE have shown neuroprotective effects with use alone and as a complement to TH. A review in 2012 did not reveal any statistically significant difference in the risk of death or a composite of death or severe neurodevelopmental disability.
Currently, the ALBINO trial, a European double-blinded randomized placebo-controlled parallel group multicenter trial (Phase III), is evaluating the effect of postnatal allopurinol administered in addition to standard of care (including therapeutic hypothermia if indicated) on the incidence of death and severe neurodevelopmental impairment at 24 months of age in newborns with perinatal hypoxic–ischemic insult and signs of potentially evolving encephalopathy [66, 67].
Magnesium sulfate
Magnesium sulfate is an NMDA receptor antagonist with two therapeutic effects (antiexcitotoxic and antiapoptotic) that is widely used antenatally for neuroprotection in preterm deliveries. A prospective, longitudinal, placebo-controlled trial of MgSO4 use in infants with severe asphyxia, without hypothermia therapy, demonstrated good short-term outcomes compared to standard supportive treatment [68]. A systematic review of preclinical evidence for MgSO4 use in HIE demonstrated no benefit and little consensus in dose and timing of administration [69]. Another meta-analysis evaluating MgSO4 in HIE concluded that there was improvement in short-term outcomes and no increase in side effects [70]. However, further large studies are needed to determine if there are long-term benefits of magnesium and to confirm its safety.
Stem cells
Different studies recently suggested that after an HI event, there is increase of regeneration pathways, opening up new research into therapies not only to attenuate brain damage but also to promote cell repair and regeneration in a developmentally disorganized brain long after the perinatal insult [71, 72]. Exogenous stem cell transplantation for neonatal HIE shows promising preliminary results. The delivery routes for stem cell transplantation include intracerebral, intrathecal, arterial, intravenous, intraperitoneal, and intranasal approaches.
Stem cell-based therapy has the potential to rescue and replace the ischemic tissue caused by HI and may facilitate endogenous brain repair [73]. The implantantion of neural precursor cells (derived from embryonic stem cells) into the deep motor cortex of HIE newborn rats can improve motor function, within 3 weeks post-implantation [74].
Intranasal delivery of human neural stem cells (HNSC) could improve neurobehavioral outcomes in neonatal HI rats, which is possibly related to the modulation of NF-κB signaling [75].
The noninvasive nasal cavity implantation provides a simple method to perform stem cell transplantation in the future. The clinical application of HNSC has been preliminarily attempted in infants with HIE. Luan et al. [76] reported that HNSC were transplanted into one 75-day old male infant with neurological disability that was caused by severe HIE; 28 days after transplantation, remarkable improvement occurred not only in his myotonia but also in his intelligence and movement, which became similar to those of the normal infants of the same age. Positron emission tomography (PET) showed significantly increased radioactivity at temporal and occipital lobes which suggested that the cellular metabolism had increased greatly.
In another study, HNSC were implanted in six cases of neonatal HIE; the second day after cell transplantation, all patients’ sucking and swallowing reflexes appeared, convulsions stopped, and muscle tension was improved. All patients were evaluated at 12 months of age; four cases showed normal mental motor development, whereas two cases presented with cerebral palsy [77]. The above studies have provided very valuable experience for further research and clinical applications.
Cord blood mononuclear cells
Cord blood cell transplantation may be the most potent therapeutic candidate for neonatal HIE, with wide application prospects.
These cells show a strong potential for nerve regeneration and they are easier to obtain than other stem cells. Animal experiments have demonstrated that cord blood mononuclear cells can reduce the activity of microglia, inhibit neuronal cell death, and promote the recovery of sensorimotor reflex function [78]. Also, they can promote the differentiation of endogenous neural stem cells into mature neurons to aid in the recovery and development of damaged neurons [79] and the upregulation of neurotrophic factor in the brain after cell transplantation [80]. The optimal timing, dosage, and delivery of stem cell transplantation for neonatal HIE will require further study.
Conclusions
HIE is the result of severe anoxic brain injury during neonatal period that can lead to premature mortality or can result in life-long morbidity.
Nowadays, the only recognized treatment for neonatal HIE is hypothermia that, although positively influencing the neurological outcome of neonatal HIE [81], is only partially effective. In fact, no more than 50% of the neonates treated achieve an improved outcome [11, 82].
At the present, efforts are focusing on finding adjuvant therapies for HIE and understanding the pathophysiology of HIE is the first, indispensable, step to better clarify the mechanism underlying it.
Pathophysiology of HIE involves several different events, strictly linked one to the others. Mitochondrial dysfunction, excitotoxicity, calcium surge, reactive oxygen species accumulation, and inflammation are the five main events caused by severe anoxic brain injury. Starting from them, a variety of complex pathways begins. Some gaps in our knowledge concerning the pathophysiology and the timing of important endogenous neuroprotective and neuroregenerative mechanisms still exist.
Although there have been significant steps in the basic sciences to create novel neuroprotective and intervention strategies to combat HIE, there is still much more research needed to be conducted to translate potential life-saving and brain damage-limiting therapies.
Abbreviations
- AIF:
-
Apoptosis-inducing factor
- BBB:
-
Blood–brain barrier
- BIP:
-
Bax-inhibiting peptide
- ETC:
-
Electron transport chain
- HI:
-
Hypoxia–ischemia
- HIE:
-
Hypoxic–ischemic encephalopathy
- HNSC:
-
Human neural stem cells
- MMPs:
-
Matrix metalloproteinases
- MPTP:
-
Mitochondrial permeability transition pore
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NO:
-
Nitric oxide
- PARP:
-
Poly(ADP-ribose) polymerase
- PDHC:
-
Pyruvate dehydrogenase complex
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- TGF-β:
-
Transforming growth factor beta
- TNF-α:
-
Tumor necrosis factor alpha
References
Adstamongkonkul D, Hess DC (2017) Ischemic conditioning and neonatal hypoxic ischemic encephalopathy: a literature review. Cond Med 1(1):9–16
Shah P, Perlman M (2009) Time courses of intrapartum asphyxia: neonatal characteristics and outcomes. Am J Perinatol 26(1):39–44
Kurinczuk J, White-Koning M, Badawi N (2010) Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 86:329–338
Lawn J, Shibuya K, Stein C (2005) No cry at birth: global estimates of intrapartum stillbirths and intrapartum-related neonatal deaths. Bull World Health Organ 83(6):409–417
Montaldo P, Pauliah SS, Lally PJ, Olson L, Thayyil S (2015) Cooling in a low-resource environment: lost in translation. Semin Fetal Neonatal Med. 20(2):72–79. https://doi.org/10.1016/j.siny.2014.10.004
Favié LMA, Cox AR, van den Hoogen A, Nijboer CHA, Peeters-Scholte CMPCD, van Bel F, Egberts TCG, Rademaker CMA, Groenendaal F (2018) Nitric oxide synthase inhibition as a neuroprotective strategy following hypoxic–ischemic encephalopathy: evidence from animal studies. Front Neurol 9:258. https://doi.org/10.3389/fneur.2018.00258
Wassink G, Gunn ER, Drury PP, Bennet L, Gunn AJ (2014) The mechanisms and treatment of asphyxial encephalopathy. Front Neurosci 8:40. https://doi.org/10.3389/fnins.2014.00040
Liu F, McCullough LD (2013) Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin 34:1121–1130
Higgins RD, Raju T, Edwards AD et al (2011) Hypothermia and other treatment options for neonatal encephalopathy: an executive summary of the Eunice Kennedy Shriver NICHD workshop. J Pediatr 159:851–858
Edwards AD, Brocklehurst P, Gunn AJ et al (2010) Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 340:c363
Carloni S, Facchinetti F, Pelizzi N, Buonocore G, Balduini W (2018) Melatonin acts in synergy with hypothermia to reduce oxygen-glucose deprivation-Induced cell death in rat hippocampus organotypic slice cultures. Neonatology 114:364–371
Marcelino T, de Lemos RP, Miguel P, Netto C, Pereira Silva L, Matte C (2015) Effect of matermal exercise on biochemical parameters in rats submitted to neonatal hypoxia-ischemia. Brain Res 1622:91–101
Edwards AB, Anderton RS, Knuckey NW, Meloni BP (2018) Perinatal hypoxic-ischemic encephalopathy and neuroprotective peptide therapies: a case for cationic arginine-rich peptides (CARPs). Brain Sci. https://doi.org/10.3390/brainsci8080147
Gunn A, Laptook A, Robertson N, Barks J, Thoresen M, Wassink GA (2017) Therapeutic hypothermia translates from ancient history in to practice. Pediatr Res 81:202–209
Zubrow ABDPM, Ashraf Q, Fritz K, Mishra O (2002) Nitric oxide-mediated Ca2+/calmodulin-dependent protein kinase IV activity during hypoxia in neuronal nuclei from newborn piglets. Neurosci Lett 335:5–8
Cao W, Carney J, Duchon A, Floyd R, Chevion M (1988) Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci Lett 88(2):233–238
Zubrow AB, Delivoria-Papadopoulosm M, Ashrafm QM et al (2002) Nitric oxide-mediated expression of Bax protein and DNA fragmentation during hypoxia in neuronal nuclei from newborn piglets. Brain Res 954(1):60–67
Groenendaal F, Vles J, Lammers H, De Vente J, Smit D, Nikkels P (2008) Nitrotyrosine in human neonatal spinal cord after perinatal asphyxia. Neonatology 93(1):1–6
Dorrepaal C, van Bel F, Moison R, Shadid M, van de Bor M, Steendijk P et al (1997) Oxidative stress during post-hypoxic-ischemic reperfusion in the newborn lamb: the effect of nitric oxide synthesis inhibition. Pediatr Res 41:321–326
Lu Y, Tucker D, Dong Y, Zhao N, Zhuo X, Zhang Q (2015) Role of mitochondria in neonatal hypoxic-ischemic brain injury. J Neurosci Rehabil 2(1):1–14
Erecinska M, Silver I (1994) Ions and energy in mammalian brain. Prog Neurobiol 43(1):37–71
Gilland E, Puka-Sundvall M, Hillered L, Hagberg H (1998) Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature brain: involvement of NMDA receptors. J Cereb Blood Flow Metab 18(3):297–304
Wang K (2000) Calpain and caspase: can you tell the difference? Trends Neurosci 23(1):20–6
Wang H, Pathan N, Ethell I, Krajewski S, Yamaguchi Y, Shibasaki F et al (1999) Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 229:339–343
McDonald J, Johnston M (1990) Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15:41–70
Johnston MV, Trescher WH, Ishida A, Nakajima W, Zipursky A (2001) Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr Res 49(6):735–741
Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328(2):309–316
Thornton C, Rousset CI, Kichev A, Miyakuni Y, Vontell R, Baburamani AA et al (2012) Molecular mechanisms of neonatal brain injury. Neurol Res Int 2012:e506320
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LGD, Neumann D et al (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13(22):2004–2008
Concannon CG, Tuffy LP, Weisová P et al (2010) AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol 189(1):83–94. https://doi.org/10.1083/jcb.200909166
Uchino H, Minamikawa-Tachino R, Kristián T, Perkins G, Narazaki M, Siesjö B et al (2002) Differential neuroprotection by cyclosporin A and FK506 following ischemia corresponds with differing abilities to inhibit calcineurin and the mitochodrial permeability transition. Neurobiol Dis 10(3):219–233
Alessandri B, Rice A, Levasseur J, DeFord M, Hamm R, Bullock M (2002) Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma 19(7):829–841
Perier C, Tieu KGC, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A et al (2005) Complex I deficiency primes bax dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci USA 102(52):19126–21913
Thornton C, Hagberg H (2015) Role of mitochondria in apoptotic and necroptotic cell death in the developing brain. Clin Chim Acta 451:35–38
Nakajima W, Ishida ALM, Gabrielson K, Wilson M, Martin L, Blue M et al (2000) Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci 20(21):7994–8004
Montaldo P, Kaforou M, Pollara G et al (2019) Whole blood gene expression reveals specific transcriptome changes in neonatal encephalopathy. Neonatology 115(1):68–76
Northington FJ, Chavez-Valdez R, Martin LJ (2011) Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol 69(5):743–758
Ginet V, Puyal J, Clarke PG, Truttmann AC (2009) Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol 175(5):1962–1974
Koike M, Shibata M, Tadakoshi M et al (2008) Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol 172(2):454–469
Xu Y, Tian Y, Tian Y, Li X, Zhao P (2016) Autophagy activation involved in hypoxic-ischemic brain injury induces cognitive and memory impairment in neonatal rats. J Neurochem 139(5):795–805
Choi D, Rothman SM (1990) The role of glutamate neurotoxicity in hypoxicischemic neuronal death. Ann Rev Neurosci 13:171–182
Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17:796–808
Del Bigio M, Becker L (1994) Microglial aggregation in the dentate gyrus: a marker of mild hypoxic-ischaemic brain insult in human infants. Neuropathol Appl Neurobiol 20:144–151
Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS et al (2015) The role of inflammation in perinatal brain injury. Nat Rev Neurol 11:192–208
Faustino J, Wang X, Johnson C, Klibanov A, Derugin N, Wendland M et al (2011) Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci 31:12992–13001
Varnum M, Ikezu T (2012) The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp 60:251–266
Swanson R, Ying W, Kauppinen T (2004) Astrocyte influences on ischemic neuronal death. Curr Mol Med 4:193–205
Anderson C, Swanson R (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32:1–14
Jiang N, Chopp M, Chahwala S (1998) Neutrophil inhibitory factor treatment of focal cerebral ischemia in the rat. Brain Res 788:25–34
Bona E, Andersson A, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K et al (1999) Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res 45(4 Pt 1):500–509
Yıldız E, Ekici B, Tatlı B (2017) Neonatal hypoxic ischemic encephalopathy: an update on disease pathogenesis and treatment. Expert Rev Neurother 17(5):449–459
Liu J, Feng ZC (2010) Increased umbilical cord plasma interleukin-1 beta levels was correlated with adverse outcomes of neonatal hypoxicischemic encephalopathy. J Trop Pediatr 56:178–182
Green H, Treacy E, Keohane A, Sullivan A, O’Keeffe G, Nolan Y (2012) A role for interleukin-1beta in determining the lineage fate of embryonic rat hippocampal neural precursor cells. Mol Cell Neurosci 49:311–312
Martin D, Chinookoswong N, Miller G (1994) The interleukin-1 receptor antagonist (rhIL-1ra) protects against cerebral infarction in a rat model of hypoxia-ischemia. Exp Neurol 130:362–367
Baggiolini M (2001) Chemokines in pathology and medicine. J Intern Med 250:91–104
Wu Q, Chen W, Sinha B et al (2015) Neuroprotective agents for neonatal hypoxic-ischemic brain injury. Drug Discov Today 20(11):1372–1381
Nair J, Kumar VHS (2018) Current and emerging therapies in the management of hypoxic ischemic encephalopathy. Children (Basel). https://doi.org/10.3390/children5070099
Faulkner S, Bainbridge A, Kato T, Chandrasekaran M, Kapetanakis AB, Hristova M, Liu M, Evans S, De Vita E, Kelen D et al (2011) Xenon augmented hypothermia reduces early lactate/N-acetylaspartate and cell death in perinatal asphyxia. Ann Neurol 70:133–150
Chakkarapani E, Dingley J, Liu X, Hoque N, Aquilina K, Porter H, Thoresen M (2010) Xenon enhances hypothermic neuroprotection in asphyxiated newborn pigs. Ann Neurol 68:330–341
Azzopardi D, Robertson NJ, Bainbridge A, Cady E, Charles-Edwards G, Deierl A, Fagiolo G, Franks NP, Griffiths J, Hajnal J et al (2016) Moderate hypothermia within 6 h of birth plus inhaled xenon versus moderate hypothermia alone after birth asphyxia (TOBY-Xe): a proof-of-concept, open-label, randomised controlled trial. Lancet Neurol 15:145–153
Faulkner SD, Downie NA, Mercer CJ, Kerr SA, Sanders RD, Robertson NJ (2012) A xenon recirculating ventilator for the newborn piglet: developing clinical applications of xenon for neonates. Eur J Anaesthesiol 29:577–585
Broad KD, Fierens I, Fleiss B, Rocha-Ferreira E, Ezzati M, Hassell J, Alonso-Alconada D, Bainbridge A, Kawano G, Ma D et al (2016) Inhaled 45–50% argon augments hypothermic brain protection in a piglet model of perinatal asphyxia. Neurobiol Dis 87:29–38
Aly H, Elmahdy H, El-Dib M, Rowisha M, Awny M, El-Gohary T, Elbatch M, Hamisa M, El-Mashad AR (2015) Melatonin use for neuroprotection in perinatal asphyxia: a randomized controlled pilot study. J Perinatol 35:186–191
Wu YW, Mathur AM, Chang T, McKinstry RC, Mulkey SB, Mayock DE, Van Meurs KP, Rogers EE, Gonzalez FF, Comstock BA et al (2016) High-dose erythropoietin and hypothermia for hypoxic-ischemic encephalopathy: a phase ii trial. Pediatrics 137:e20160190
Baserga MC, Beachy JC, Roberts JK, Ward RM, DiGeronimo RJ, Walsh WF, Ohls RK, Anderson J, Mayock DE, Juul SE et al (2015) Darbepoetin administration to neonates undergoing cooling for encephalopathy: a safety and pharmacokinetic trial. Pediatr Res 78:315–322
Chaudhari T, McGuire W (2012) Allopurinol for preventing mortality and morbidity in newborn infants with hypoxic-ischaemic encephalopathy. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD006817.pub3
Maiwald CA, Annink KV, Rüdiger M et al (2019) Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (ALBINO): study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). BMC Pediatr 19(1):210
Bhat MA, Charoo BA, Bhat JI, Ahmad SM, Ali SW, Mufti MU (2009) Magnesium sulfate in severe perinatal asphyxia: a randomized, placebo-controlled trial. Pediatrics 123:e764–e769
Galinsky R, Bennet L, Groenendaal F, Lear CA, Tan S, van Bel F, Juul SE, Robertson NJ, Mallard C, Gunn AJ (2014) Magnesium is not consistently neuroprotective for perinatal hypoxia-ischemia in term-equivalent models in preclinical studies: a systematic review. Dev Neurosci 36:73–82
Tagin M, Shah PS, Lee KS (2013) Magnesium for newborns with hypoxic-ischemic encephalopathy: a systematic review and meta-analysis. J Perinatol 33:663–669
Felling RJ, Snyder MJ, Romanko MJ, Rothstein RP, Ziegler AN, Yang Z et al (2006) Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci 26(16):4359–4369
Parry SM, Peeples ES (2018) The impact of hypoxic-ischemic brain injury on stem cell mobilization, migration, adhesion, and proliferation. Neural Regen Res 13(7):1125–1135. https://doi.org/10.4103/1673-5374.235012
Daadi MM, Davis AS, Arac A et al (2010) Human neural stem cell grafts modify microglial response and enhance axonal sprouting in neonatal hypoxic–ischemic brain injury. Stroke 41(3):516–523
Shinoyama M, Ideguchi M, Kida H et al (2013) Cortical region-specific engraftment of embryonic stem cell derived neural progenitor cell restores axonal sprouting to a subcortical target and achieves motor functional recovery in a mouse model of neonatal hypoxic ischemic brain injury. Front Cell Neurosci 7:128
Ji G, Liu M, Zhao XF et al (2015) NF-kB signaling is involved in the effects of intranasally engrafted human neural stem cells on neurofunctional improvements in neonatal rat hypoxic–ischemic encephalopathy. CNS Neurosci Ther 21(12):926–935
Luan Z, Yin GC, Hu XH et al (2005) Treatment of an infant with severe neonatal hypoxic–ischemic encephalopathy sequelae with transplantation of human neural stem cell into cerebral ventricle. Zhonghua Er Ke Za Zhi 43(8):580–583 (discussion 580)
Luan Z, Liu WP, Qu SQ et al (2011) Treatment of newborns with severe injured brain with transplantation of human neural precursor cell. Zhonghua Er Ke Za Zhi 49(6):445–449
Pimentel-coelho PM, Magalhaes ES, Lopes LM et al (2010) Human cord blood transplantation in a neonatal rat model of hypoxic–ischemic brain damage: functional outcome related to neuroprotection in the striatum. Stem Cells Dev 19(3):351–358
Huang HZ, Wen XH, Liu H et al (2013) Human umbilical cord blood mononuclear cell transplantation promotes long-term neurobehavioral functional development of newborn SD rats with hypoxic ischemic brain injury. Zhonghua Er Ke Za Zhi 51(6):460–466
Yasuhara T, Hara K, Maki M et al (2010) Mannitol facilitates neurotrophic factor up-regulation and behavioural recovery in neonatal hypoxic–ischaemic rats with human umbilical cord blood grafts. J Cell Mol Med 14(4):914–921
Shankaran S (2012) Therapeutic hypothermia for neonatal encephalopathy. Curr Treat Options Neurol 14:608–619
Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG (2013) Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 1:CD003311. https://doi.org/10.1002/14651858.CD003311.pub3
Funding
There is no funding source.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Greco, P., Nencini, G., Piva, I. et al. Pathophysiology of hypoxic–ischemic encephalopathy: a review of the past and a view on the future. Acta Neurol Belg 120, 277–288 (2020). https://doi.org/10.1007/s13760-020-01308-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-020-01308-3