
Abstract
A series of novel substituted 2-(4-bromophenyl)-4-phenylthiazole compounds 5a–h were synthesized by a simplified two-step process. First, the selective and effective α-bromination of substituted acetophenone moieties with N-bromo succinamide was done followed by treating of substituted 2-bromo-1-phenylethan-1-one with 4-bromobenzothiamide under microwave irradiation. The structures of the newly synthesized compounds were confirmed by IR, 1H NMR, 13C NMR, mass spectra and C, H, N analysis. Further, all the synthesized compounds 5a–h were evaluated for xanthine oxidase inhibition. Among all the tested compounds, 5f was found to be highly potent (IC50 = 0.100 ± 0.08 µM) followed by 5e (IC50 = 0.145 ± 1.42 µM), compared to the standard allopurinol (IC50 = 0.150 ± 0.07 µM), which is evident from in vitro and in silico analysis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Xanthine oxidase (XO) is an oxidoreductase enzyme responsible for generating highly reactive oxygen species, which catalyzes the oxidation of hypoxanthine to xanthine and further catalyzes the oxidation of xanthine to uric acid [1]. Uric acid accumulation leads to gout attacks and development of kidney stones. This enzyme has been even reported to play a potential role in the progression and development of various cancers which makes it an imperative therapeutic target [2]. The ideal xanthine oxidase inhibitor (XOI) allopurinol has been the root of the clinical management of gout and conditions correlated with hyperuricemia for quite a few decades. Many people are hypersensitive to allopurinol drugs as they may develop rashes, fever, eosinophilia, hepatitis and worsened renal function [3]. Allopurinol is one of the drugs commonly known to cause Stevens–Johnson syndrome and toxic epidermal necrolysis which are life-threatening dermatological conditions. Being allergic to allopurinol is mainly due to the generation of superoxides [4, 5]. Reactive oxygen species (ROS) are formed continuously in the human body, and they are removed by enzymatic and non-enzymatic antioxidative defense systems under normal conditions [6]. ROS contribute to the oxidative stress on the organisms, and they are involved in many pathological processes such as causing damage to DNA, protein and lipids and also ensuing in inflammation, atherosclerosis, cancer and aging [7].
Thiazole is considered a synthetically significant active scaffold that possesses the majority of biological activities [8, 9]. In the current scenario, the synthesis of phenyl thiazole compounds has been a fascinating field in therapeutic science which prompts biological investigation to assess their potential pharmacological significance, because of their potent and significant biological activities [10, 11]. Thiazole and its derivatives are found to be influential in various forms of drugs. Currently, drugs which are available in the market are sulfathiazole (antimicrobial drug), ritonavir (antiretroviral drug), abafungin (antifungal drug), tiazofurin (antineoplastic drug) [12], ceftazidime [13] (broad-spectrum third-generation cephalosporin antibiotic), sudoxicam (anti-inflammatory drug) and famotidine (antiulcerogenic) [14]. Therefore, the synthesis of these compounds which shows a broad spectrum of biological activity is of remarkable concern and has attracted the attention of many researchers to explore thiazole derivatives [15, 16]. Current studies have shown that the number of cases of gout is increasing worldwide [17, 18] and more research is focused on the synthesis of non-purine-based xanthine oxidase inhibitor. US Food and Drug Administration (USFDA) for the first time approved febuxostat a non-purine drug as XO inhibitor in the year 2009 which in turn is a thiazole derivative [19]; since then, multitudinous non-purine inhibitors have attracted worldwide attention. This drug is an oral, potent and selective XO inhibitor with minimum side effects as compared with a purine drug allopurinol. The success of this prime, non-purine XO inhibitor has fascinated researchers to develop a structurally diverse array of molecules without purine skeleton [20, 21]. Owing to this importance, an approach was made to design and synthesis XO inhibitor containing thiazole moiety. In addition, the effort should fit conveniently to be carried out within a laboratory working period. In order to do this, a simple procedure is required to be developed. One such method is the microwave technique that can be safely used for solventless reactions and an opportunity to work with open vessels, thus avoiding the risk of high-pressure development. Besides, there is a possibility to upscale the reaction on a preparative scale and the reactions appear to occur at a relatively low bulk temperature [22]. In view of the above-mentioned facts and our initial efforts to discover potentially active new non-purine-based XO inhibitory agents 2-(4-bromophenyl)-4-phenylthiazole derivatives 5a–h was synthesized in good yield.
Results and discussion
Chemistry
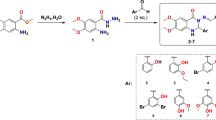
The reaction sequence for different title compounds 5a–h is outlined in Scheme 1. Each stage in the synthesis was confirmed by IR, NMR and mass spectral information. The compounds substituted 2-bromo-1-phenylethan-1-one 3a–h were synthesized according to a reported procedure [23] through selective α-bromination of substituted acetophenone 1 with N-bromosuccinimide (NBS) 2 catalyzed by p-toluenesulfonic acid (PTSA) under microwave irradiation for 25 to 30 min. To confirm the formation of the series 3a–h, compound 3a was selected as a representative. The 1H NMR spectrum of compound 3a has shown a singlet peak at δ 4.56 ppm for two methylene protons and also the disappearance of the peak for methyl protons of compound 1. Also, the broad range of the mass spectrum gave significant stable m/z 277 (M+), 279 (M+2), 281 (M+4) peaks. Further, substituted 2-bromo acetophenone 3a–h were treated with 3-bromo thiobenzamide 4 and irradiated at 350W in the microwave for 15 to 20 min to furnish 2-(4-bromophenyl)-4-phenylthiazole derivatives 5a–h with excellent yield. In this series compound, 5a was considered as a representative. The 1H NMR spectrum of compound 5a has shown peaks in the range 7.12–7.76 ppm as multiplet for aromatic protons and at δ 8.57 as singlet for thiazole ring proton. The broad range of the mass spectrum gave significant stable m/z 394 (M+), 396 (M+2), 433 (M+4) peaks. To interrogate the transformations between microwave irradiation and the thermal heating, synthesis of compounds 3a–h and 5a–h using thermostat oil-bath reflux condition otherwise identical conditions as those employed for the microwave-assisted method was studied (Tables 1 and 2). Lower yields with a longer reaction time were observed under thermal conditions, signifying that the microwave irradiation conditions offer much better efficacy.

Synthesis of 2-(4-bromophenyl)-4-phenylthiazole analogs under microwave irradiation
Enzymatic assay
Novel 2-(4-bromophenyl)-4-phenylthiazole analogs 5a–h were screened for the bovine XO. Enzyme activity was determined by measuring the absorbance of the liberated uric acid from xanthine at 294 nm using a microplate reader (Spectramax 340, Molecular Devices, Sunnyvale, USA). The absorbance was compared with the control, containing DMSO instead of test samples, and allopurinol was used as a positive control [18]. Further, on to recognize the binding mode of the newly synthesized compounds 5a–h with XO, molecular docking studies for the most potent compound 5f were carried out.
Structure–activity relationship
Substituted 2-(4-bromophenyl)-4-phenylthiazoles 5a–h, which are known to be pharmacologically active molecules, were efficiently synthesized from moderate to good yield. Structurally, the title compounds 5a–h has a basic backbone of phenyl thiazole ring. In order to get an insight into the structure–activity relationship (SAR), we varied the substitutions on the phenyl ring attached to the thiazole moiety. The IC50 values for compounds as depicted in Table 3 inferred that the compound 5f with the nitro group at a para position to the phenyl ring showed (IC50 = 0.100 ± 0.08 µM) followed by 5e with fluoro group at para position (IC50 = 0.145 ± 1.42 µM), compared to the standard allopurinol (IC50 = 0.150 ± 0.07 µM). The results illustrated that compound 5f having a thiazole ring with a nitro group at the para position of the phenyl ring is important for biological activity which was further confirmed by in silico analysis. The other compounds 5a, 5b, 5c, 5d, 5g and 5h have not shown significant activity, and the results are represented in Table 3. With this inference, the compound 5f was identified as a lead bioactive molecule.
Molecular docking simulation
To get a better understanding of the interaction between XO with the ligands, the docking simulation was performed where allopurinol was used as the standard drug. The docked result with their binding affinity and their non-bonding interaction is shown in Table 4. Further, the summary of their interaction protein along with its interaction residues, type of bond formed and their respective distance is given in Table 5. The active site was taken based on the reported work [24]. The interaction of the complex was studied using Biovia Discovery Studio Visualizer 2021. Based on the criteria as mentioned in the “methods” section, ligand 2-(4-bromophenyl)-4-(4-nitrophenyl) thiazole 5f bound to XO was chosen which showed the binding affinity of − 9.1 kJ/mol for which allopurinol was used as a control which showed the binding affinity of − 7.0 kJ/mol. On comparing both, docked model ligand compound 5f bound to XO was found to be more stable as it showed more negative binding affinity. Similarly, the compound 5f complex contains a total of four hydrogen bonds, where all four were bounded within the active site—ARG A: 880 (2.83), ARG A: 880 (2.03), THR A: 1010 (2.52) and PHE A: 1009 (2.24) similar to the active site of the co-crystal ligand as mention in the previous work done by the authors. Further, the analysis showed that the compound 5f bound to XO contains a total of ten hydrophobic bonds, which were formed between the residues of the protein, where all the residues were within the active site. The control drug allopurinol bound XO has a smaller number of total non-bonded interactions. Furthermore, the allopurinol complex showed an unfavorable bond which was formed with ALA A: 1079. Thus, it can be said that the chosen compound 5f complex is stable compared to the control drug (Figs. 1 and 2).

Visualization of docking interaction of 2-(4-bromophenyl)-4-(4-nitrophenyl)thiazole (5f) bound xanthine oxidase (PDB: 1N5X). A 3D binding interaction. B 2D residue-wise interaction

Visualization of docking interaction of allopurinol bound xanthine oxidase (PDB: 1N5X). A 3D binding interaction. B 2D residue-wise interaction
Molecular dynamics (MD) simulation
The docked complex with the most negative binding affinity was selected for future investigation to understand the dynamic nature and the conformational stability between both protein and ligand complexes. The simulation was carried out for 100 ns using GROMACS-2018.1 package for compound 5f bound XO as well as for the control drug allopurinol bound XO. The trajectories were obtained after the completion of the MD simulation. The evaluation of these trajectories provided an in-depth understanding of the complex flexibility and their stable nature during the interaction. The evaluation is done by analyzing root mean square deviation (RMSD), root means square fluctuation (RMSF), radius of gyration (RG), solvent accessible surface area (SASA) and hydrogen bonds. The RMSD is evaluated to get a clear understanding of positional differences of the backbone atoms over a timescale.
As shown in Fig. 3, the RMSD for compound 5f bound XO along with allopurinol bound XO complex was analyzed to evaluate their stability as plotted in Fig. 3A. The graphical plot of RMSD predicts that XO shows a slight upward fluctuation as time increases, and at around 50 ns, it attains stability and it shows variability between 0.27 and 0.28 nm. Later, it attains stability around 80 ns for the run of 100 ns. Further, RMSD of compound 5f complex shows stability in fluctuation from 55 to 70 ns, but there is a slight fluctuation at 0.31 nm around 60 ns and equilibrated around 80 ns during 100 ns while allopurinol complex shows consistencies in stability at ~ 15 nm. The RMSF plot was analyzed to evaluate the residue flexibility of the complex along with the reference ligand complex as shown in Fig. 3B. Thorough fluctuation of the protein was found to be higher compared to the protein–ligand complexes. The RG was analyzed to evaluate the structural compactness of the biomolecules and to check whether the complexes are stably folded or unfolded. The average RG value of XO was 3.01 nm, but it showed a slight increase in the fluctuation value (3.14 nm) and later showed relatively stable values among the analyzed complexes. Compound 5f complex showed average RG value of 3.13 nm by showing more or less stable values compared to allopurinol complex of 3.15 nm. The protein, as well as complexes, showed almost similar and compactable values as shown in Fig. 3C. So, the above evaluation indicates that the complexes may be showing relatively stable folded conformation during MD simulation. Figure 3D shows the graphical plot of SASA through which conformational changes between the interactions are analyzed. The overall SASA value of the protein and both the complexes is consistently similar. It can also be seen that there is a slight downgrade fluctuation of compound 5f complex at 440 nm2 which is similar to allopurinol. Thus, the evaluated analyses show that there can be compactness of the structure which may lead to increase in stability. Finally, the number of hydrogen bonds that were formed during the interaction with both the modeled protein were calculated, where compound 5f bound XO showed up to four hydrogen bonds in Fig. 3E, whereas allopurinol bound XO showed three hydrogen bonds. There is no variation in hydrogen bonds formed in transformation from docked model to MD simulated model which shows that there may not be much structural rearrangement.

Analysis of RMSD, RMSF, Rg, SASA and number of hydrogen bonds of of 2-(4-bromophenyl)-4-(4-nitrophenyl)thiazole 5f (black) and allopurinol (violet) bound to xanthine oxidase (red) (PDB: 1N5X) at 100 ns. A Time evolution of backbone RMSD of the complex structure. B RMSF of protein and ligand. C Radius of gyration (Rg). D SASA. E Hydrogen bonds occurring over the time of simulation between protein and ligand
Binding free energy calculation
The binding free energy was evaluated using the molecular mechanics/Poisson–Boltzmann surface area (MM-PBSA) approach which is the summation of non-polar, polar and non-bonded interaction energies. It is known that the more the negative energy, the more the stability, and based on this concept, the value was evaluated. The summary of the calculation of the binding energy of van der Waal energy, electrostatic energy, polar solvation and free binding energy for 100 ns is given in Table 6 for compound 5f bound XO as well as for the control drug allopurinol bound XO, with its values and standard deviation. It is seen that for compound 5f bound xanthine oxidase, van der Waals (− 226.854 kJ/mol), electrostatic interactions (− 24.635 kJ/mol), SASA energy (− 17.105 kJ/mol) negatively contribute to the total interaction energy, whereas polar solvation energy (108.105) positively accords to the total interaction free energy and the binding energy predicted by the program was − 106.488 kJ/mol. It can be seen that the polar solvation was not favorable for the binding of the compound. While the control drug allopurinol showed the binding energy − 25.906 kJ/mol, van der Waals −55.892 kJ/mol, electrostatic interactions − 3.951 kJ/mol, SASA energy − 3.706 kJ/mol similar to compound 5f polar solvation energy, allopurinol polar solvation energy also has a positive value 37.643 kJ/mol. The evidence shows that van der Waals, electrostatic interaction and non-polar interaction together accord to complex stability compared to standard drug. Furthermore, when the standard deviation of analog 5f bound XO was compared with the control drug, it showed more stability as the values were slower to mean. Similarly, compared to the control drug, 5f showed more stability as the value predicted by the program was more negative.
Conclusion
Novel 2-(4-bromophenyl)-4-phenylthiazole analogs were efficiently synthesized from microwave irradiation. Compound 5f having a thiazole ring with a nitro group at the para position of the phenyl ring is confirmed potent inhibitory activity against XO which was further confirmed by in silico analysis. The XO inhibitory potential of test compounds 5a–h is shown in Table 1. In vitro XOI studies revealed that all the compounds exhibited good XO inhibitory activity in the range of 0.100 ± 0.08 to 0.418 ± 1.10 µM. Among them, compound 5f was found to be highly potent (IC50 = 0.100 ± 0.08 µM) followed by 5e (IC50 = 0.145 ± 1.42 µM). The remaining compounds showed lower activity compared to the standard allopurinol (IC50 = 0.150 ± 0.07 µM). Further, molecular docking simulation analysis showed that the compound 5f bound to XO contains a total of ten hydrophobic bonds, where all the residues were within the active site. The control drug allopurinol bound to XO has a smaller number of non-bounded interactions. MD simulations accounted that there is not much structural rearrangement from docked model to MD simulated model transformation as compound 5f bound to XO showed up to four hydrogen bonds, whereas allopurinol bound to XO showed three hydrogen bonds. Binding free energy calculation predicted that, compared to the control drug, compound 5f showed more stability as the value predicted by the program was more negative. In conclusion, our in vitro and in silico analysis for compound 5f gave strong evidence that proved it to be a better XO inhibitor compared to the standard drug allopurinol.
Materials and methods
Chemicals were purchased from Sigma-Aldrich Chemical Co. TLC was done on aluminum backed silica plates and visualized by UV light. Solvents were dried and degassed according to “Purification of Laboratory Chemicals” prior to use. Melting points were determined on a Thomas Hoover capillary melting point apparatus with a digital thermometer. The FT-IR spectra were recorded using KBr disks and Nujol on FT-IR Jasco 4100 infrared spectrophotometer, 1H NMR and 13C NMR spectra were recorded on a Bruker 400 MHz NMR spectrophotometer in CDCl3, and the chemical shifts were recorded in parts per million downfield from tetramethylsilane. Mass spectra were recorded on LCMS/MS (API4000) mass spectrometer. The elemental analysis of the compounds was performed on a PerkinElmer 2400 elemental analyzer. The results of elemental analyses remained within ± 0.4% of the theoretical values. The reactions were performed in a CEM discover microwave reactor.
Chemistry
General procedure for the synthesis of substituted 2-bromo-1-phenylethan-1-one (3a–h)
A 10-mL dry flask with a teflon stir bar was introduced with substituted acetophenone 1a–h (0.0129 mol), NBS (0.0129 mol, 1 equiv) and PTSA (0.00129 mol, 10 mol%). Anhydrous dichloro methane (2.0 mL) was added; then, the flask was sealed and the mixture was stirred and irradiated at 180w under microwave. After 25 to 30 min, the reaction mixture was cooled and treated with 10 mL of distilled water, and extracted with 3 × 10 mL of dichloro methane. The organic layers were separated and dried over anhydrous magnesium sulfate, and the solution was concentrated using rotavapour under high pressure and the resulting solid was crystallized in ethanol to afford desired compounds 3a–h in a pure state.
2-Bromo-1-(2-chloro-4-nitrophenyl)ethan-1-one 3a: IR (Nujol): 1658 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.56 (s, 2H), 8.10–8.28 (m, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 31.11 (1C, CH2), 121.92 (1C, Ar–C), 124.82 (1C, Ar–C), 131.32 (1C, Ar–C), 134.54 (1C, Ar–C), 144.64 (1C, Ar–C), 157.54 (1C, Ar–C), 190.82 (1C, C=O). MS: m/z 277(M+), 279 (M+2), 281 (M+4). Anal. Calcd. for C8H5BrClNO3 (277): C, 34.50; H, 1.81; N, 5.03. Found: C, 34.48; H, 1.79; N, 5.00%.
2-Bromo-1-(2-fluoro-4-nitrophenyl)ethan-1-one 3b: IR (Nujol): 1668 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.63 (s, 2H), 8.09–8.76 (m, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 31.11 (1C, CH2), 111.92 (1C, Ar–C), 119.42 (1C, Ar–C), 131.42 (1C, Ar–C), 131.74 (1C, Ar–C), 153.64 (1C, Ar–C), 160.74 (1C, Ar–C), 190.82 (1C, C=O). MS: m/z 261(M+), 263 (M+2). Anal. Calcd. for C8H5BrFNO3 (261): C, 36.67; H, 1.92; N, 5.35. Found: C, 36.60; H, 1.89; N, 5.33%.
2-Bromo-1-(4-chloro-2-fluorophenyl)ethan-1-one 3c: IR (Nujol): 1648 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.56 (s, 2H), 7.37–7.89 (m, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 31.06 (1C, CH2), 117.63 (1C, Ar–C), 124.69 (1C, Ar–C), 127.82 (1C, Ar–C), 131.33 (1C, Ar–C), 140.40 (1C, Ar–C), 164.68 (1C, Ar–C), 190.48 (1C, C=O). MS: m/z 251 (M+), 253 (M+2), 255 (M+4). Anal. Calcd. for C8H5BrClFO (251): C, 41.15; H, 2.00; Found: C, 41.11; H, 2.00%.
2-Bromo-1-(4-chlorophenyl)ethan-1-one 3d: IR (Nujol): 1639 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.47 (s, 2H), 7.63 (dd, 2H), 8.03 (dd, 2H). 13C NMR (125 MHz, CDCl3) δ (ppm): 30.99 (1C, CH2), 128.7 (2C, Ar–C), 130.2 (2C, Ar–C), 132.3 (1C, Ar–C), 138.7 (1C, Ar–C), 190.8 (1C, C=O). MS: m/z 233 (M+), 235 (M+2), 237 (M+4). Anal. Calcd. for C8H6BrClO (233): C, 41.15; H, 2.59; Found: C, 41.11; H, 2.57%.
2-Bromo-1-(4-fluorophenyl)ethan-1-one 3e: IR (Nujol): 1659 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.41 (s, 2H), 7.40 (dd, 2H), 8.12 (dd, 2H). 13C NMR (125 MHz, CDCl3) δ (ppm): 30.13 (1C, CH2), 115.65 (2C, Ar–C), 129.90 (1C, Ar–C), 130.33 (2C, Ar–C), 166.78 (1C, Ar–C), 189.99 (1C, C=O). MS: m/z 217 (M+), 219 (M+2). Anal. Calcd. for C8H6BrFO (217): C, 44.27; H, 2.79; Found: C, 44.24; H, 2.76%.
2-Bromo-1-(4-nitrophenyl)ethan-1-one 3f: IR (Nujol): 1668 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.46 (s, 2H), 8.31 (dd, 2H), 8.40 (dd, 2H). 13C NMR (125 MHz, CDCl3) δ (ppm): 29.77 (1C, CH2), 123.69 (2C, Ar–C), 129.72 (2C, Ar–C), 137.94 (1C, Ar–C), 150.29 (1C, Ar–C), 189.52 (1C, C=O). MS: m/z 243(M+), 245 (M+2). Anal. Calcd. for C8H6BrNO3 (243): C, 39.37; H, 2.49; Found: C, 39.34; H, 2.46%.
2-Bromo-1-(4-bromophenyl)ethan-1-one 3 g: IR (Nujol): 1638 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.40 (s, 2H), 7.78 (dd, 2H), 7.95 (dd, 2H). 13C NMR (125 MHz, CDCl3) δ (ppm): 30.01(1C, CH2), 128.97(2C, Ar–C), 130.06 (2C, Ar–C), 131.86(1C, Ar–C), 132.21(1C, Ar–C), 190.07 (1C, C = O). MS: m/z 277 (M+), 279 (M+2), 281 (M+4). Anal. Calcd. for C8H6Br2O (277): C, 34.57; H, 2.19; Found: C, 34.54; H, 2.16%.
2-Bromo-1-(4-bromo-2-chlorophenyl)ethan-1-one 3 h: IR (Nujol): 1643 cm–1 (C=O). 1H NMR (400 MHz, CDCl3) δ (ppm): 4.53 (s, 2H), 7.61–7.83 (m, 3H). 13C NMR (125 MHz, CDCl3) δ (ppm): 31.01 (1C, CH2), 128.69 (1C, Ar–C), 129.82 (1C, Ar–C), 130.33 (1C, Ar–C), 132.40 (1C, Ar–C), 135.74 (1C, Ar–C), 137.68 (1C, Ar–C), 190.82 (1C, C=O). MS: m/z 312 (M+), 314 (M+2), 316 (M+4), 318 (M+6). Anal. Calcd. for C8H5Br2ClO (312): C, 30.76; H, 1.61; Found: C, 30.73; H, 1.59; N, 5.00%.
General procedure for the synthesis of substituted 2-(4-bromophenyl)-4-phenylthiazoles (5a–h)
In a 25 ml dry flask with a teflon stir bar, substituted 2-bromo-1-phenylethan-1-one 3a–h (0.00214 mol) containing 15 ml of ethanol were introduced and it was followed by the addition of 3-bromo thiobenzamide 4 (0.00214 mol, 1 equiv) and irradiation at 350W in the microwave for 15 to 20 min. The solid was washed with diethyl ether and followed with water and brine solution. The organic layer was separated using a separating funnel. Further, it was dried with anhydrous magnesium sulfate, and the solution was concentrated using rotavapour under high pressure and the resulting solid was crystallized in ethanol to afford desired compounds 5a–h in a pure state.
2-(4-Bromophenyl)-4-(2-chloro-4-nitrophenyl) thiazole 5a: Yield 89%, m.p. 145.3–147.2 °C. IR (Nujol): 3159 cm−1 (Ar–C–H) str, 1647 cm−1 (C=N) str, 1593 and 1325 cm−1 asymmetric and symmetric (N=O) str of NO2, 1507 cm−1 (Ar–C=C) str, 1109 cm−1 (C–N) str, 667 cm−1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 7.12–7.76 (m, 7H, Ar–H), 8.57 (s, 1H, thiazole). 13C NMR (125 MHz, CDCl3) δ (ppm): 111.59 (1C, thiazole), 122.59 (1C, Ar–C), 124.73 (1C, Ar–C), 129.70 (1C, Ar–C), 129.77 (2C, Ar–C), 132.63 (2C, Ar–C), 133.25 (1C, Ar–C), 136.86 (1C, Ar–C), 149.34 (1C, Ar–C), 158.27 (1C, thiazole), 164.89 (1C, thiazole). MS: m/z 394 (M+), 396 (M+2), 433 (M+4). Anal. calcd. for C15H8BrClN2O2S (394): C, 45.54; H, 2.04; N, 7.08. Found: C, 45.52; H, 2.04; N, 7.05%.
2-(4-Bromophenyl)-4-(2-fluoro-4-nitrophenyl)thiazole 5b: Yield 82%, m.p. 137.3–138.7 °C. IR (Nujol): 3159 cm−1 Ar–(C–H) str, 1647 cm–1 (C=N) str, 1593 and 1325 cm–1 asymmetric and symmetric (N=O) str of NO2, 1507 cm–1 Ar–(C=C) str, 1109 cm–1 (C–N) str, 667cm-1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 7.07–8.22 (m, 7H, Ar–H), 8.23 (s, 1H, thiazole). 13C NMR (125 MHz, CDCl3) δ (ppm): 111.59 (1C, thiazole), 117.9.59 (1C, Ar–C), 120.63 (1C, Ar–C), 129.64 (1C, Ar–C), 129.6 (1C, Ar–C), 129.77 (2C, Ar–C), 130.63 (1C, Ar–C), 132.25 (2C, Ar–C), 142.86 (1C, Ar–C), 149.54 (1C, Ar–C), 153.27 (1C, thiazole), 159.2 (1C, Ar–C), 163.89 (1C, thiazole). MS: m/z 378 (M+), 380 (M+2). Anal. calcd. for C15H8BrFN2O2S (378): C, 47.51; H, 2.13; N, 7.39. Found: C, 47.49; H, 2.14; N, 7.36%.
2-(4-Bromophenyl)-4-(4-chloro-2-fluorophenyl)thiazole 5c: Yield 87%, m.p. 142.3–145.5 °C. IR (Nujol): 3159 cm−1 Ar–(C–H) str, 1647 cm−1 (C=N) str, 1507 cm−1 Ar–(C=C) str, 1109 cm−1 (C–N) str, 667 cm−1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 6.89–7.99 (m, 7H, Ar–H), 8.03 (s, 1H, thiazole). 13C NMR (125 MHz, CDCl3) δ (ppm): 111.54 (1C, thiazole), 118.10 (1C, Ar–C), 121.43 (1C, Ar–C), 123.62 (1C, Ar–C), 124.65 (1C, Ar–C), 129.72 (2C, Ar–C),130.57 (1C, Ar–C), 132.13 (2C, Ar–C), 135.9 (1C, Ar–C), 142.94 (1C, Ar–C), 153.97 (1C, thiazole), 159.72 (1C, Ar–C), 162.84 (1C, thiazole). MS: m/z 367 (M+), 369 (M+2). 371 (M+4). Anal. calcd. for C15H8BrClFNS (367): C, 48.87; H, 2.19; N, 3.80. Found: C, 48.86; H, 2.15; N, 3.79%.
2-(4-Bromophenyl)-4-(4-chlorophenyl)thiazole 5d: Yield 80%, m.p. 93.4–95.4 °C. IR (Nujol): 3153 cm−1 Ar–(C–H) str, 1641 cm−1 (C=N) str, 1502 cm−1 Ar–(C=C) str, 1107 cm−1 (C–N) str, 663cm–1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 6.89 (dd, J = 9 Hz, 2H, Ar–H), 7.25 (dd, J = 4 Hz, 2H, Ar–H), 7.27 (dd, J = 4 Hz, 2H, Ar–H), 7.68 (dd, J = 8 Hz, 2H, Ar–H), 8.02 (s, 1H, thiazole). 13C NMR (125 MHz, CDCl3) δ (ppm): 111.52 (1C, thiazole), 123.10 (1C, Ar–C), 128.92 (2C, Ar–C), 129.73 (2C, Ar–C), 129.42 (2C, Ar–C),131.07 (1C, Ar–C), 132.74 (2C, Ar–C), 134.34 (1C, Ar–C), 142.34 (1C, Ar–C), 153.29 (1C, thiazole), 164.84 (1C, thiazole). MS: m/z 351 (M+), 353 (M+2), 355 (M+4). Anal. calcd. for C15H9BrClNS (351): C, 51.38; H, 2.59; N, 3.99. Found: C, 51.35; H, 2.57; N, 3.98%.
2-(4-Bromophenyl)-4-(4-fluorophenyl)thiazole 5e: Yield 89%, m.p. 121.4–123.5 °C. IR (Nujol): 3159 cm−1 Ar–(C–H) str, 1647 cm−1 (C=N) str, 1507 cm−1 Ar–(C=C) str, 1109 cm−1 (C–N) str, 667 cm−1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 7.51 (dd, J = 8 Hz, 2H, Ar–H), 7.76 (dd, J = 8 Hz, 2H, Ar–H), 7.84 (dd, J = 9 Hz, 2H, Ar–H), 8.10 (dd, J = 8 Hz, 2H, Ar–H), 9.04 (s, 1H, thiazole). 13C NMR (125 MHz, CDCl3) δ (ppm): 111.54 (1C, thiazole), 116.10 (2C, Ar–C), 128.92 (2C, Ar–C), 123.73 (1C, Ar–C), 129.72 (1C, Ar–C),130.07 (2C, Ar–C), 132.14 (2C, Ar–C), 142.34 (1C, Ar–C), 153.19 (1C, thiazole), 162.94 (1C, Ar–C), 165.64 (1C, thiazole). MS: m/z 333 (M+), 335 (M+2). Anal. calcd. for C15H9BrFNS (333): C, 53.91; H, 2.71; N, 4.91. Found: C, 53.88; H, 2.71; N, 4.90%.
2–(4–Bromophenyl)–4–(4–nitrophenyl)thiazole 5f: Yield 92%, m.p. 132.2–135.5 °C. IR (Nujol): 3159 cm−1 Ar–(C–H) str, 1647 cm−1 (C=N) str, 1593 and 1325 cm−1 asymmetric and symmetric (N=O) str of NO2, 1507 cm−1 Ar–(C=C) str, 1109 cm−1 (C–N) str, 667cm−1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 7.36 (dd, J = 8 Hz, 2H, Ar–H), 7.44 (dd, J = 8 Hz, 2H, Ar–H), 7.57 (dd, J = 16 Hz, 2H, Ar–H), 7.99 (dd, J = 8 Hz, 2H, Ar–H), 8.24 (s, 1H, thiazole, 13C NMR (125 MHz, CDCl3) δ (ppm): 104.54 (1C, thiazole), 116.10 (2C, Ar–C), 119.43 (1C, Ar–C), 122.32 (2C, Ar–C), 125.62 (1C, Ar–C), 127.42 (2C, Ar–C), 130.17 (2C, Ar–C), 144.23 (1C, Ar–C), 152.59 (1C, Ar–C), 153.24 (1C, Ar–C), 158.19 (1C, thiazole), 160.89 (1C, thiazole). MS: m/z 360 (M+), 362 (M+2). Anal. calcd. for C15H9BrN2OS (360): C, 49.88; H, 2.50; N, 7.76. Found: C, 49.87; H, 2.49; N, 7.74%.
2,4-Bis(4-bromophenyl)thiazole 5 g: Yield 89%, m.p. 113.2–117.5 °C. IR (Nujol): 3159 cm−1 Ar–(C–H) str, 1647 cm−1 (C=N) str, 1507 cm−1 Ar–(C=C) str, 1109 cm−1 (C–N) str, 667 cm−1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 7.46 (dd, J = 24 Hz, 4H, Ar–H), 7.96 (dd, J = 16 Hz, 4H, Ar–H), 7.98 (s, 1H, thiazole). 13C NMR (125 MHz, CDCl3) δ (ppm): 111.54 (1C, thiazole), 116.10 (2C, Ar–C), 128.32 (2C, Ar–C), 123.83 (1C, Ar–C), 129.62 (1C, Ar–C),130.17 (2C, Ar–C), 132.23 (2C, Ar–C), 142.34 (1C, Ar–C), 153.19 (1C, thiazole), 162.94 (1C, Ar–C), 164.89 (1C, thiazole). MS: m/z 395 (M+), 397 (M+2). Anal. calcd. for C15H9Br2NS (395): C, 45.60; H, 2.30; N, 3.55. Found: C, 45.58; H, 2.30; N, 3.53%.
2-(4-Bromophenyl)-4-(4-bromo-2-chlorophenyl)thiazole 5 h: Yield 87%, m.p.135.3–137.2 °C. IR (Nujol): 3159 cm−1 Ar–(C–H) str, 1647 cm−1 (C=N) str, 1507 cm−1 Ar–(C=C) str, 1109 cm−1 (C–N) str, 667 cm−1 (C–S) str. 1H NMR (CDCl3) δ (ppm): 6.90–7.59 (m, 7H, Ar–H), 8.05 (s, 1H, thiazole). 13C NMR (125 MHz, CDCl3) δ (ppm): 111.57 (1C, thiazole), 123.10 (1C, Ar–C), 124.53 (1C, Ar–C), 128.90 (1C, Ar–C), 129.70 (2C, Ar–C), 130.25 (1C, Ar–C), 130.86 (1C, Ar–C), 131.19 (1C, Ar–C), 132.13 (2C, Ar–C), 134.43 (1C, Ar–C), 143.94 (1C, Ar–C), 153.27 (1C, thiazole), 162.84 (1C, thiazole). MS: m/z 429 (M+), 431 (M+2), 433 (M+4). Anal. calcd. for C15H8Br2ClNS (429): C, 41.94; H, 1.88; N, 3.26. Found: C, 4.86; H, 2.15; N, 3.79%.
Enzymatic assay
Xanthine oxidase inhibition assay
The bovine XO (EC 1.17.3.2., categorized as xanthine: oxygen oxidoreductase) inhibition was assayed using the substrate xanthine as per the modified method described [18]. In brief, 700 µl phosphate buffer (50 mM, pH 7.4) and 100 µl of test samples with diverse concentrations dissolved in DMSO (dimethyl sulfoxide) were mixed prior to the addition of 100 µl of bovine xanthine oxidase (0.25 U/ml). The assay mixture was pre-incubated for 10 min at 30 °C. After incubation, 100 µl of 1.0 mM xanthine solution in water was added and the reaction was maintained at 30 °C for 10 min. Enzyme activity was determined by measuring the absorbance of the liberated uric acid from xanthine at 294 nm using a microplate reader (Spectramax 340, Molecular Devices, Sunnyvale, USA). The absorbance was compared with the control, containing DMSO instead of test samples. Allopurinol was used as a positive control. The results were expressed as percent xanthine oxidase inhibition attained using the formula given below:
Each experiment was performed in triplicates, along with appropriate blanks. The concentration required to inhibit 50% of xanthine oxidase activity under the specified assay conditions was described as the IC50.
Molecular docking simulation
The crystal structure of xanthine dehydrogenase (PDB: 1N5X) [21] was obtained from the protein data bank (https://www.rcsb.org/) which has a resolution of 2.8 Å. The preparation of both the target protein and the ligand was done following autodock 4.2 protocols [25]. Prior to the docking process, water molecules and hetero atoms were removed. The target protein was added with sufficient atoms of hydrogen to balance the structure. Further, to this, both the charges of Kollman united and Gasteiger–Marsili empirical atomic partial charges were added followed by assigning of the AD4 atom type to the macromolecule. The two-dimensional structure of the ligand was drawn using ChemSketch 2020 1.2 for which 3D optimization was done. The 3d structure which was drawn was converted to the autodock desired file format using OpenBabel 2.3.1 [26]. The preparation of the ligand was done similarly to the target protein, where the default charges were kept for Kollman united and Gasteiger. The docking simulation was performed based on structure-based technique which is most reliable. The grid box of 20 × 20 × 20 Å and the coordinates of x = 96.663 Å, y = 54.963 Å and z = 39.433 Å were created within the active site [24]. The docking process was done according to autodock Vina 1.1.2 protocol [27]. The results obtained after docking process contain a total of nine poses for each docked complex. The best pose was chosen based on RMSD, binding energy, the total number of non-bonded interactions and respective conventional as well as carbon hydrogen bonds. The result was analyzed using Biovia Discovery Studio Visualizer 2021. Finally, the chosen docked model was selected to study its dynamic nature.
Molecular dynamics simulation
The docked complex of the chosen model with the most negative binding affinity and RMSD with less than 1.0 Å and most hydrogen bonds is used for the molecular dynamic study. The MD simulation is carried out using the GROMACS-2018.1 [28] for the duration of 100 ns, which is a biomolecular software package. These studies were carried out by applying the CHARMM27 force field. The water molecules of TIP3 were applied for the protein–ligand complex. The ligand topology was obtained using SwissParam server (https://www.swissparam.ch/) [29]. The entire simulation was carried on at nanoseconds scale for which the pdb2gmx module was used. The solvent box of 10 Å distance was created followed by vacuum minimization of 5000 steps using the steepest descent algorithm. The neutralization of the entire system was done by adding counter ions (Na+ and Cl−) as well as by maintaining the suitable salt concentration of 0.15 M which was followed by energy minimization using the steepest descent algorithm for 5000 steps. Constant temperature and pressure were maintained throughout the MD simulation. Finally, before MD production, system was equilibrated in NVT and subsequent NPT ensemble (1000 ps each) with a 310 K temperature and 1 bar pressure. The trajectories were analyzed for RMSD, RMSF, Rg, SASA and hydrogen bonds, and plotted using the XMGRACE program [30].
Binding free energy calculation
By using the MM-PBSA approach the free binding energy of the chosen complex was calculated, for which g_mmpbsa program tool which works based on GROMACS-2018.1 is used [31]. The calculation is done using MD trajectories of the last 50 ns considered to compute ΔG with dt 1000 frames. It is evaluated using molecular mechanical energy, polar and apolar solvation energies. The Eqs. (1 and 2) to calculate the free binding energy are given below.
ADMET and drug likeliness studies
The absorption, distribution, metabolism, excretion and toxicity (ADMET) screening was carried out to know their drug likeliness, pharmacokinetics parameters like absorption, distribution, metabolism, elimination properties as well as their toxicity through in silico approach using vNN-ADME and OSIRIS property explorer (http://www.cheminfo.org/Chemistry/Cheminformatics/Property_explorer) [32, 33]. The drug likeliness was evaluated based on “rule of five,” similarly the toxicity was evaluated based on mutagenic, tumorigenic, irritant, effects on the reproductive system, drug-induced liver injury (DILI) and cytotoxicity. All the above parameters were predicted with the available databases which contain available drug compounds.
Drug likeliness and pharmacokinetics analyses
The “rule of five” of Lipinski’s and the pharmacokinetic potential such as absorption, distribution, metabolism, elimination properties were studied for analog 5f with control drug, allopurinol. The drug score of analog 5f predicted by OSIRIS was 0.76. Similarly, for allopurinol, the drug score predicted by OSIRIS was 0.68. The pharmacokinetic property was analyzed based on their molecular weight (352.15 g/mol), number of hydrogen bond acceptors (4), number of rotatable bonds (3) and cLogP (3.87). Likewise, there was no toxicity found and drug administration predicted by databases showed P-glycoprotein (P-gp) and cytochrome P (CYP) inhibition. Further, the in silico evaluation of mutagenicity, tumorigenicity, irritability showed negative on the compound.
References
V. Lakshmi Ranganatha, A. Bushra Begum, P. Naveen, F. Zameer, R. Hegdekatte, S.A. Khanum, Synthesis, xanthine oxidase inhibition, and antioxidant screening of benzophenone tagged thiazolidinone analogs. Arch. Pharm. Chem. Life Sci. 347, 589–598 (2014)
B.A. Bushra, B. Muneera, R.V. Lakshmi, T. Prashanth, Z. Farhan, H. Raghavendra, A.K. Shaukath, Synthesis, antioxidant and xanthine oxidase inhibitory activity of 5-[4-[2-(5-Ethyl-2-pyridinyl) ethoxy] phenyl] methyl]-2, 4-thiazolidinedione derivatives. Arch. Pharm. Chem. Life Sci. 347, 247–255 (2014)
W.H. Chung, C.W. Wang, R.L. Dao, Severe cutaneous adverse drug reactions. J. Dermatol. 43, 758–766 (2016)
H.D. Gurupadaswamy, V. Girish, F. Zameer, R. Hegdekatte, J.B. Chauhan, S.A. Khanum, Synthesis of pyrimidones and evaluation of their xanthine oxidase inhibitory and anti-oxidant activities. Arch. Pharm. Chem. Life Sci. 346, 805–811 (2013)
C.E. Berry, J.M. Hare, Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J. Physiol. 555, 589–606 (2004)
L.D. Kong, Y. Zhang, X. Pan, R.X. Tan, C.H.K. Cheng, Inhibition of xanthine oxidase by liquiritigenin and isoliquiritigenin isolated from sinofranchetia chinensis. Cell. Mol. Life Sci. 57, 500–505 (2000)
B. Halliwell, J.M.C. Gutteridge, C.E. Cross, Free radicals, antioxidants, and human disease. J. Lab. Clin. Med. 119, 598–620 (1992)
T. Prashanth, B.R. Vijay Avin, P. Thirusangu, V. Lakshmi Ranganatha, B.T. Prabhakar, J.N. Narendra Sharath Chandra, S.A. Khanum, Synthesis of coumarin analogs appended with quinoline and thiazole moiety and their apoptogenic role against murine ascitic carcinoma. Biomed. Pharmacother. 112, 108707 (2019)
Y.H.E. Mohammed, V.H. Malojirao, P. Thirusangu, M. Al-Ghorbani, B.T. Prabhakar, S.A. Khanum, The novel 4-phenyl-2-phenoxyacetamide thiazoles modulates the tumor hypoxia leading to the crackdown of neoangiogenesis and evoking the cell death. Eur. J. Med. Chem. 143, 1826–1839 (2018)
H.A. Khamees, Y.H.E. Mohammed, S. Ananda, F.H. Al-Ostoot, Y. Sangappa, S. Alghamdi, S.A. Khanum, M. Madegowda, Effect of o-difluoro and p-methyl substituents on the structure, optical properties and anti-inflammatory activity of phenoxy thiazole acetamide derivatives theoretical and experimental studies. J. Mol. Struct. 1199, 127024 (2020)
M. Al-Ghorbani, H.A. Alghamdi, S.A. Khanum, A review on anticancer potential of thiazole-heterocyclic hybrid compounds. Eur. J. Biomed. Pharm. Sci. 5, 122–128 (2018)
N. Siddiqui, M.F. Arshad, W. Ahsan et al., Thiazoles: a valuable insight into the recent advances and biological activities. Int. J. Pharm. Sci. Drug Res. 1, 136–143 (2009)
M.S. Masoud, A.E. Ali, N.M. Nasr, Chemistry, classification, pharmacokinetics, clinical uses, and analysis of beta-lactam antibiotics: a review. J Chem Pharm Res. 6, 58 (2014)
M.A.M.A. Shaikh, M. Raghuwanshi, M. Khurshid, S. Nazim, A.A.A. Rafique, Schiff’s bases and amides of selected five membered heterocyclic compounds: a review. J. Chem. Pharm. Res. 5, 14–25 (2013)
H.A. Khamees, Y.H.E. Mohammed, A. Swamynayaka, F.H. Al-Ostoot, Y. Sert, S. Alghamdi, S.A. Khanum, M. Madegowda, Molecular structure, DFT, vibrational spectra with fluorescence effect, hirshfeld surface, docking simulation and antioxidant activity of thiazole derivative. Chem. Select. 4, 4544–4558 (2019)
P. Thirusangu, V. Vigneshwaran, T. Prashanth, B.R. VijayAvin, V.H. Malojirao, H. Rakesh, S.A. Khanum, R. Mahmood, B.T. Prabhakar, Angiogenesis BP-1T, an antiangiogenic benzophenone-thiazole pharmacophore, counteracts HIF-1 signalling through p53/MDM2-mediated HIF-1a proteasomal degradation. Angiogenesis 1, 55–71 (2017)
H.M. Kramer, G. Curhan, The association between gout and nephrolithiasis: the national health and nutrition examination survey III, 1988–1994. Am. J. Kidney Dis. 40, 37–42 (2002)
K.R. Sathisha, S.A. Khanum, J.N. Narendra Sharath Chandra, F. Ayisha, S. Balaji, K. Gopal Marathe, G. Shubha, K.S. Rangappa, Bioorg. Med. Chem. 19, 211–220 (2010)
M.K. Reinders, T.L. Jansen, Management of hyperuricemia in gout: focus on febuxostat. Clin. Interv. Aging. 05, 7–18 (2010)
G. Kaur, J.V. Singh, M.K. Gupta, K. Bhagat, H.K. Gulati, A. Singh, P.M.S. Bedi, H. Singh, S. Sharma, Med. Chem. Res. 29, 83–93 (2020)
M.R. Ali, S. Kumar, O. Afzal, N. Shalmali, M. Sharma, S. Bawa, Xanthine oxidase inhibitory activity of aminothiazole. Chem. Biol. Drug Des. 87, 508–516 (2016)
S.A. Khanum, S. Shashikanth, B.S. Sudha, Microwave-assisted synthesis of 2-amino and 2-azetidinonyl-5-(2-benzoyl-phenoxymethyl)-1,3,4-oxadiazoles. Heteroat. Atom Chem. 15, 37 (2004)
X.-Y. Guan, Z. Al-Misbaa, K.-W. Huang, Arab. J. Chem. 8, 892–896 (2015)
K. Okamoto, B.T. Eger, T. Nishino, S. Kondo, E.F. Pai, T. Nishino, An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J. Biol. Chem. 278, 1848–1855 (2003)
G.M. Morris, R. Huey, W. Lindstrom, M.F. Sanner, R.K. Belew, D.S. Goodsell, A.J. Olson, AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009)
N.M. O’Boyle, M. Banck, C.A. James, C. Morley, T. Vandermeersch, G.R. Hutchison, Open babel: an open chemical toolbox. J. Cheminf. 03, 1–4 (2011)
O. Trott, A.J. Olson, AutoDock Vina, improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010)
M.J. Abraham, T. Murtola, R. Schulz, S. Páll, J.C. Smith, B. Hess, E. Lindahl, High performance molecular simulations through multi-level parallelism from laptops to supercomputers. GROMACS SoftwareX 1, 19–25 (2015)
S.V. Zoete, M.A. Cuendet, A. Grosdidier, O. Michielin, Swiss param, a fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 (2011)
P.J. Turner, XMGRACE, Version 5.1. 19. Center for Coastal and Land-Margin Research (Oregon Graduate Institute of Science and Technology, Beaverton, OR, 2005)
R. Kumari, R. Kumar, A GROMACS tool for high-throughput MM-PBSA calculations g_mmpbsa. Open-source drug discovery consortium, lynn A. J. Chem. Inf. Model. 54, 1951–1962 (2014)
B. Sander, A. Nizam, L.P. Garrison Jr., M.J. Postma, M.E. Halloran, Economic evaluation of influenza pandemic mitigation strategies in the United States using a stochastic microsimulation transmission model. J. Natl. Lib. Med. 12, 226–233 (2009)
P. Schyman, R. Liu, V. Desai, A. Wallqvist, vNN web server for ADMET predictions. J. Front. Pharmacol. 4, 889 (2017)
Acknowledgements
All authors thank the University of Mysore, Mysuru, and JSS Academy of Higher Education and Research, Mysuru, for providing facilities to carry out the biological activity. Shaukath Ara Khanum thankfully acknowledges the financial support provided by VGST, Bangalore, under CISEE Programme [Project sanction order: No. VGST/CISEE/282].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Rights and permissions
About this article

Cite this article
Jyothi, M., Khamees, H.A., Patil, S.M. et al. Microwave-Assisted Synthesis, Characterization, Docking Studies and Molecular Dynamic of Some Novel Phenyl Thiazole Analogs as Xanthine Oxidase Inhibitor. J IRAN CHEM SOC 19, 3919–3933 (2022). https://doi.org/10.1007/s13738-022-02574-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02574-z