
Abstract
Several novel quinazoline derivatives bearing phenolic hydroxyl moiety (2–7) have been produced with good yields and screened for biological activities. All the title compounds were characterized using spectroscopic techniques such as 1H NMR, 13C NMR, FTIR, and HRMS. Then, the anti-browning effects of synthesized quinazoline derivatives were investigated in vitro. The IC50 values for molecules 2–7 were calculated as 0.085, 1.145, 0.106, 6.86, 0.52, 7.07 µM, respectively. Ki constants, which are inhibitory-enzyme binding constants, were calculated by using Lineweaver–Burk graphs as 0.16 ± 0.0620, 0.906 ± 0.3029, 0.055 ± 0.0171, 9.363 ± 2.5809, 0.773 ± 0.3204, 7.863 ± 1.9107 µM, respectively. In computer-aided analysis, to gain insights electrochemical properties, synthesized compounds were analysed theoretically by density functional theory. Molecular docking studies and MD simulations were performed to identify possible inhibitor-enzyme binding interactions. According to obtained results, all the compounds formed hydrogen bonds with Asn 112 and Asn 414, and showed π-cation interaction with Phe2 70, gatekeeper residue in target protein. Supporting the nm level inhibition, MD simulations indicate that protein-inhibitor complex maintain the stability and have high number of hydrogen bond formation during the simulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polyphenols continue to be a current topic for researchers because of their wide range antioxidant, antimicrobial and anti-inflammatory effects (Contente et al. 2021). A increasing number of epidemiological studies have related the consumption of polyphenol-rich fruits and vegetables to the protection of chronic diseases, particularly cardiovascular disease, neurodegenerative disorders, and some specific cancers (Scalbert et al. 2005; Khurana et al. 2013). They also have various positive impact on plasma, membranes, transcription factors and enzyme activities (Li et al. 2014).
The quinazoline nucleus, a fascinating molecule among the most significant groups of aromatic heterocyclic compounds with two nitrogen atoms in its structure, represents a uniquely diversified class of frameworks with diverse biological activities (Kumar et al. 2022). Compounds belonging to this intriguing class are of significant interest in biotechnological and medical applications. They serve as 'privileged motifs' for drug discovery due to their affinity for numerous receptors (Khan et al. 2016). Derivatives with various substituents demonstrate a broad spectrum of biological activities, including antibacterial (Raghavendra et al. 2007; Panneerselvam et al. 2009), antimalarial (Verhaeghe et al. 2008), anti-inflammatory (Alagarsamy et al. 2009; Saravanan et al. 2010; Smits et al. 2010), anticonvulsant (Georgey et al. 2008; Patel et al. 2010), antihypertensive (Ismail et al. 2006), antioxidant (Ahmad 2017), antiviral (Krishnan et al. 2011), anti-HIV25 (Pati and Banerjee 2013), and anticancer (Nerkar et al. 2009; Manasa et al. 2011; Sak 2012; Ahmed and Youns 2013).
Browning in fruits and vegetables results in a deterioration of their organoleptic qualities and nutritional properties, ultimately leading to product rejection by consumers (Mathew and Parpia 1971; Queiroz et al. 2008).
Polyphenoloxidase (PPO), also known as tyrosinase, polyphenolase, phenolase, catechol oxidase, cresolase, and catecholase, constitutes the primary enzyme group responsible for the enzymatic browning process (Mayer 2006). PPO is activated in the processes such as slicing and cutting fruits and vegetables, and formation of brown pigments starts immediately (Kuijpers et al. 2012). This enzyme exhibits two distinct reaction mechanisms in the presence of molecular oxygen: hydroxylation of monophenols to o-diphenols (monophenolase activity), and oxidation of o-diphenols to o-quinones (Tinello and Lante 2018).
The control of enzymatic browning holds paramount importance in the food industry. While various physical and chemical methods are employed for this purpose, ongoing efforts continue to introduce novel strategies. Traditional chemical approaches to prevent browning in post-harvest crops primarily rely on the inhibition of polyphenol oxidase (McEvily et al. 1992). PPO inhibitors are divided into four groups according to their mechanism of action: reducing agents, acid regulators, complexing agents and chelating agents (Kuijpers et al. 2012; Kuijpers et al. 2013; Liu et al. 2013; Guiamba and Svanberg 2016). Previous studies have shown that the use of conventional chemical inhibitors such as citric acid (Zhou et al. 2016), ascorbic acid (Siddiq and Dolan 2017), L-cysteine (Siddiq and Dolan 2017) or sulphur compounds (Kuijpers et al. 2012) have significant inhibitory effects on PPO activity.
In light of the ever-evolving challenges posed by enzymatic browning in various food products, the current research and development of new, effective polyphenol oxidase (PPO) inhibitors are valuable for the food industry. Accordingly, this study concerns synthesizing new polyphenols containing quinazolinone ring and to investigate their inhibitory effects on PPO enzyme in vitro to find more potent PPO inhibitors. We also provide in current paper an in-depth computational calculation using molecular docking and molecular dynamics that may aid future studies on the design of new quinazoline derivatives.
Experimental
Synthesis
The chemicals used in the synthesis were purchased from various suppliers. Melting points were determined on WRS-2A Microprocessor Melting-point Apparatus and are uncorrected. The FTIR spectra of the compounds were recorded using ALPHA-P BRUKER FTIR spectrophotometer. 1H NMR spectras were recorded on Bruker (400 MHz) spectrometer. 13C NMR spectras were recorded on Bruker (100 MHz) spectrometer. Chemical shifts were reported as δ in ppm relative to tetramethylsilane (TMS) (δ 0.00 singlet) in deuterated chloroform (CDCl3) and deuterated dimethyl sulphoxide (DMSO-d6). HRMS spectra were recorded on Agilent 6530 Accurate-Mass spectrometer and acetonitrile was used as the solvent.
Synthesis of 2-amino-4,5-dimethoxybenzohydrazide (1)
Methyl 2-amino-4,5-dimethoxybenzoate (10 mmol) was dissolved in hydrazinium hydroxide (W/W: 80%, 10 mL), and the mixture was refluxed for an hour (monitored by TLC (Hex/EtOAc: 1/1)) without any solvent. The mixture was cooled to room temperature, and formed crystals were filtered off and washed with cold water several times. Beige crystals, yield: 90%, mp: 180–182 ℃ (lit: 182–184 ℃) [24] (Fig. 1).

Synthetic route for preparation of title compounds 1–7
Synthesis of compounds (2–7)
Aromatic aldehyde derivative (10 mmol) was dissolved in absolute ethanol (20 mL), and catalitic amount of acetic acid (3–4 drops) was added to this solution. Then, the compound 1 (5 mmol) was added and refluxed for two hours (monitored by TLC (Hex/EtOAc: 7/3)). The mixture was cooled to room temperature, and formed solid was filtered off and recrystallized from ethanol (Fig. 1).
3-(2-Hydroxybenzylideneamino)-2-(2-hydroxyphenyl)-6,7-dimethoxy-2,3-dihydroquinazolin-4(1H)-one (2)
Yellow solid, yield 83%, mp: 201–203 °C. FTIR (cm−1): νmax 3563, 3453, 3354, 3078, 2976, 1666, 1618, 1583, 1244. 1H NMR (400 MHz, DMSO) δ 11.63 (s, 1H), 10.24 (s, 1H), 8.33 (s, 1H), 7.37 (d, J = 9.3 Hz, 1H), 7.31–7.24 (m, 2H), 7.19 – 7.10 (m, 2H), 6.96–6.84 (m, 5H), 6.70 (t, J = 7.9 Hz, 1H), 6.51 (s, 1H), 3.73 (s, 6H). 13C NMR (101 MHz, DMSO) δ 160.1, 157.5, 155.0, 154.8, 148.01, 142.5, 142.1, 131.3, 130.5, 129.8, 125.9, 124.0, 119.1, 119.0, 118.4, 116.5, 115.9, 109.9, 104.6, 98.2, 65.7, 55.8, 55.4. HRMS (Q-TOF) m/z calcd for C23H21N3O5 [M + H]+: 420.1559, found: 420.1555.
3-(3-Ethoxy-2-hydroxybenzylideneamino)-2-(3-ethoxy-2-hydroxyphenyl)-6,7-dimethoxy-2,3-dihydroquinazolin-4(1H)-one (3)
Yellow solid, yield 81%, mp: 192–193 °C. FTIR (cm−1): νmax 3348, 3157, 2996, 1619, 1589, 1262. 1H NMR (400 MHz, CDCl3) δ 11.33 (s, 1H), 8.86 (s, 1H), 7.43 (s, 1H), 6.90–6.77 (m, 4H), 6.70 (d, J = 4.8 Hz, 2H), 6.50 (s, 1H), 6.18 (s, 1H), 6.09 (s, 1H), 4.13–4.06 (m, 4H), 3.85 (s, 3H), 3.81 (s, 3H), 1.47–1.42 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 161.7, 154.8, 154.7, 148.9, 147.5, 146.0, 143.3, 142.9, 141.1, 123.6, 123.5, 120.0, 118.8, 118.3, 115.5, 112.1, 110.2, 106.9, 98.2, 69.5, 64.8, 56.2, 55.9, 14.9. HRMS (Q-TOF) m/z calcd for C27H29N3O7 [M + H]+: 508.2084, found: 508.2077.
3-(5-Bromo-2-hydroxybenzylideneamino)-2-(5-bromo-2-hydroxyphenyl)-6,7-dimethoxy-2,3-dihydroquinazolin-4(1H)-one (4)
White solid, yield 87%, mp: 216–218 °C. FTIR (cm−1): νmax 3386, 3238, 3070, 2998, 1658, 1622, 1583, 1273. 1H NMR (400 MHz, DMSO) δ 11.44 (s, 1H), 10.59 (s, 1H), 8.41 (s, 1H), 7.64 (d, J = 2.5 Hz, 1H), 7.41 (dd, J = 8.8, 2.5 Hz, 1H), 7.32 (dd, J = 8.6, 2.5 Hz, 1H), 7.25 (s, 2H), 6.95 (d, J = 2.4 Hz, 1H), 6.92–6.85 (m, 2H), 6.72 (s, 1H), 6.49 (s, 1H), 3.74 (s, 6H). 13C NMR (101 MHz, DMSO) δ 160.0, 156.5, 155.1, 154.4, 146.3, 142.3, 133.6, 132.5, 131.5, 128.2, 126.5, 120.9, 118.9, 118.1, 110.2, 109.9, 109.7, 104.3, 98.2, 65.9, 55.8, 55.5. HRMS (Q-TOF) m/z calcd for C23H19Br2N3O5 [M + H]+: 575.9770, found: 575.9755.
3-(3,5-Dibromo-2-hydroxybenzylideneamino)-2-(3,5-dibromo-2-hydroxyphenyl)-6,7-dimethoxy-2,3-dihydroquinazolin-4(1H)-one (5)
White solid, yield 89%, mp: 235–237 °C. FTIR (cm−1): νmax 3281, 3058, 2960, 1647, 1592, 1239. 1H NMR (400 MHz, DMSO) δ 12.61 (s, 1H), 10.26 (s, 1H), 8.21 (s, 1H), 7.84–7.66 (m, 3H), 7.38 (s, 1H), 7.26 (s, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.89 (s, 1H), 6.50 (s, 1H), 3.75 (s, 6H). 13C NMR (101 MHz, DMSO) δ 159.5, 155.4, 153.4, 151.2, 145.8, 142.5, 141.9, 135.6, 135.0, 132.5, 129.0, 127.5, 120.9, 113.5, 111.3, 110.2, 109.5, 103.9, 98.5, 65.5, 55.8, 55.6. HRMS (Q-TOF) m/z calcd for C23H17Br4N3O5 [M + H]+: 731.7980, found: 731.7976.
3-(4-Hydroxy-3-methoxybenzylideneamino)-2-(4-hydroxy-3-methoxyphenyl)-6,7-dimethoxy-2,3-dihydroquinazolin-4(1H)-one (6)
Yellow solid, yield 84%, mp: 196–198 °C. FTIR (cm−1): νmax 3468, 3256, 3077, 2978, 1646, 1596, 1280. 1H NMR (400 MHz, DMSO) δ 9.55 (brs, 1H), 9.06 (brs, 1H), 8.62 (s, 1H), 7.38 (s, 1H), 7.25 (s, 1H), 7.17 (s, 1H), 7.13–7.04 (m, 2H), 6.82 (d, J = 8.1 Hz, 1H), 6.72–6.67 (m, 2H), 6.38 (s, 1H), 6.27 (s, 1H), 3.78–3.69 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 162.1, 155.1, 154.5, 148.1, 146.8, 146.6, 146.1, 143.8, 140.53, 131.6, 127.4, 123.2, 120.6, 114.2, 114.1, 110.3, 109.6, 109.2, 108.0, 98.7, 75.6, 56.3, 56.0, 56.0, 55.9. HRMS (Q-TOF) m/z calcd for C25H25N3O7 [M + H]+: 480.1771, found: 480.1758.
3-(4-Hydroxy-3,5-dimethoxybenzylideneamino)-2-(4-hydroxy-3,5-dimethoxyphenyl)-6,7-dimethoxy-2,3-dihydroquinazolin-4(1H)-one (7)
White solid, yield 85%, mp: 190–192 °C. FTIR (cm−1): νmax 3247, 3087, 1643, 1612, 1260. 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 7.43 (s, 1H), 6.83 (s, 2H), 6.73 (s, 2H), 6.26 (s, 1H), 6.14 (s, 1H), 3.86–3.79 (m, 18H). 13C NMR (101 MHz, CDCl3) δ 162.2, 155.1, 155.0, 154.5, 147.3, 147.2, 147.0, 143.8, 140.7, 137.2, 135.2, 130.7, 126.2, 110.3, 109.1, 105.0, 104.5, 98.7, 76.1, 56.4, 56.3, 56.1. HRMS (Q-TOF) m/z calcd for C27H29N3O9 [M + H]+: 540.1982, found: 540.1970.
Studies on polyphenol oxidase
Preparation of homogenates
Ten grams of sample of potato was ground in a pestle using the freeze–thaw method and homogenized in the extraction solution (0.05 M phosphate buffer (pH 7.4) containing 10 mM ascorbic acid, 0.5% polyethylene glycol and 1 mL % Triton X-100) by using a homogenizer for 2 min. The crude extract sample was filtered through four layers of cheesecloth. The homogenate was centrifuged at 20000xg for 30 min at 4 °C. The supernatant was collected as crude protein solution.
Purification of PPO
The synthesis of the affinity gel (Sepharose-4B-L-tyrosine p-aminobenzoic) was performed according to the method described by Arslan et al. (2004). The synthesized gel was loaded upon the affinity column preequilibrated with 0.05 M phosphate buffer (pH 6.0). The gel was washed with the same buffer after adding the enzyme extract. The elution of PPO was performed with a solution of 0.05 M phosphate buffer that contained 1 M KCl (pH 8.0).
Assay of PPO activity
PPO activity was determined spectrophotometrically by the measurement of increases in absorbance at 420 nm using catechol as substrate. One unit of PPO activity was determined as the amount of enzyme causing 0.001 increase in absorbance per minute in 1 mL of reaction mixture (Flurkey 1986).
Determination of inhibition parameters
Novel quinazoline derivates were used as PPO inhibitors and the effects of inhibitors on potato PPO activity were determined using catechol as a substrate. IC50 value was defined the inhibitor concentration that reduced 50% enzyme activity, and it was calculated with equation of Activity%-[Inhibitor (mM)] graphs. Three constant inhibitor concentrations were tested for measuring PPO activities at with five different substrate concentrations. The Lineweaver–Burk plots were drawn for each inhibitor by determining 1/V-1/[S] values at different substrate concentrations for inhibitors. In addition, Ki values and inhibition types were obtained from the graphs.
Docking analyse
3D structure of PPO (PDB ID: 6HQI) (Kampatsikas et al. 2019a, b) was downloaded from the protein data bank (https://www.rcsb.org). Preparation of enzyme comfomation was carried out with autodock tools 1.5.6 for specifying and samples rotatable bonds and computing partial charges. The active site of PPOs includes two copper ions, thus, grid box was chosen at 40 × 40 × 40 Å3 (spacing = 0.375 Å) centred these ions. Chemical structures of molecules 2–7 were drawn at ChemDraw 19.1 programme, and each of them were optimized in the gas phase using the ORCA programme, employing the Density Functional Theory (DFT) method/B3LYP functional and 6-311G (d, p) basis set. Optimized structure of compound 2 and 4 can be seen in Fig. 4. Then, prepared ligands were transferred to Avogadro (Hanwell et al. 2012). After preparations of ligands and PPO enzyme, docking analysis were carried out via the Autodock 4.2 and Autodock vina executed in the PyRx platform (Trott and Olson 2010; Dallakyan and Olson 2015). Discovery Studio Visualizer was utilized for the visualization of the 2D and 3D models of the docking conformations and interactions (Biovia and DSME 2017).
Molecular dynamics simulations
After molecular docking analyses, best-fitting pose of compound 2 in the 6hqi topologies were prepared by extracting them from the complex file generated with Pymol (DeLano 2002). Parameters for compound 2 were generated using the SwissParam webserver (Zoete et al. 2011). Protein topology was generated using the pdb2gmx tool within GROMACS and solvated in a decahedral box employing the TIP3 solvent model. 44 sodium and 35 chlorine ions were incorporated to neutralize the system. Following box boundary definition, energy minimization was conducted employing the steepest descent algorithm with 50.000 steps. Following this, a two-step equilibration process was conducted: 100 ps of NVT equilibration at 300 K using the V-rescale thermostat (Bussi, Donadio et al. 2007), and 100 ps of NPT equilibration at 1 bar using the Parrinello-Rahman barostat.
MD simulations were executed on the PPO enzyme and compound 2 a period of 200 ns for more realistic model using GROMACS 2024.1 package, implementing the CHARMM36 force field (Abraham et al. 2015). Trajectory analysis from the MD simulation was performed using GROMACS analysis tools. Root mean square deviation (RMSD) and root mean square fluctuations (RMSF) were computed using gmx rms and gmx rmsf tools, respectively. Radius of gyration (Rg) and hydrogen bond analysis were carried out using the gyrate and gmx hbond tools. All resulting files were saved with the xvg extension. Visualization and trajectory analysis to evaluate the complex's movement were performed using VMD software version 2.0.2.
DFT studies
DFT calculations were carried with Orca 5.0.2 program (Neese 2022) by using B3LYP hybrid model and 6-311G(d.p) basis set. [1.2.3]. For visualization and graphical representation of the HOMO/LUMO energies of synthesized compounds 2–7 Iboview programme was used (Knizia 2013).
Results and discussion
Synthesis
In this study, seven compounds were synthesized. Methyl-2-amino-4,5-dimethoxy benzoate was used starting compound for the synthesis of the target compounds. For this purpose, methyl-2-amino-4,5-dimethoxy benzoate was converted to its hydrazide derivative 1 by the treatment with hydrazinium hydroxide. Finally, compound 1 was reacted with the various phenolic benzaldehydes and the target polyphenols containing quinazolinone ring (2–7) were obtained with good yields (81–89%) (Fig. 1). The structures of the target novel compounds were characterized with FTIR, 1H NMR, 13C NMR, and HRMS spectroscopic methods.
In the IR spectra of the compounds (2–7), the broad stretching bands of the OH and NH groups were seen at 3563–3386 cm−1 and 3354–3238 cm−1, respectively. Aromatic and aliphatic C–H bands appeared at 3087–3058 cm−1 and 2998–2960 cm−1, respectively. C=O stretching bands of the quinazolinone ring were observed at 1666–1643 cm−1, and the CH=N stretching bands were seen at 1596–1583 cm−1. The bending bands of the N–N bands were seen at 1280–1239 cm−1. These bands are the characteristic bands and the values are fully compatible with the structures and literature (Hricovíniová et al. 2018).
In the 1H NMR spectra of the compounds (2–7), peaks of OH protons were seen as a broad singlet at δ 12.61–9.55 ppm and peaks of N=CH protons were observed as a singlet at δ 8.88–8.21 ppm. Aromatic protons were seen at δ 7.81–6.09 ppm as doublet, triplet and multiplet relative to their chemical environment. The NH protons appeared at δ 7.64–7.38 ppm and the CH protons at position 2 of the quinazolinone ring resonated at δ 6.72–6.27 ppm. Peaks of the methoxy protons were observed at δ 3.86–3.69 ppm. For compound 3, peaks of the ethoxy protons were seen as a multiplet at δ 4.13–4.06 ppm for two –CH2 and a multiplet at δ 1.47–1.42 ppm for two –CH3. Chemical shifts, integrations and splits are fully compatible with the structures and literature (Hricovíniová et al. 2018; Tokalı et al. 2021).
In the 13C NMR spectra of the new compounds (2–7), peaks of C=O carbons resonated at δ 162.2–159.5 ppm and the peaks of HC=N carbons of benzylidenamino moieties were seen at δ 155.1–145.8 ppm. Peaks of aromatic carbons were observed at δ 157.5–98.2 ppm. Peaks of the CH carbons at position 2 of the quinazolinone ring were seen at δ 65.9–64.8 ppm for the compounds 2–5 (2-OH substituted) and δ 76.1–75.6 ppm for the compounds 6 and 7 (4-OH substituted). Peaks of the methoxy carbons appeared at δ 56.4–55.4 ppm. Chemical shifts and number of the peaks are fully compatible with the structures and literature (Scalbert et al. 2005; Khurana et al. 2013).
In vitro inhibition
The exploration of strategies for inactivating the polyphenol oxidase (PPO) enzyme, which is accountable for enzymatic browning in foods, has been extensively studied across diverse sources, including apricot (eddine Derardja, Pretzler et al. 2019), apple (Janovitz-Klapp et al. 1990), grape (Fortea et al. 2009), tea leaf (Öztürk et al. 2020), potato (Aksoy 2020), lettuce (Chazarra et al. 1996), shrimp (Nirmal and Benjakul 2009). In addition, many methods have been tested for this aim, including reducing agents (Bottino, Degl’Innocenti et al. 2009), changes in pH (Zhou et al. 2020) and increasing temperature (Gouzi et al. 2012), ultrasonication (Zhu et al. 2019). Sulphites are widely used in the inactivation of the enzyme by chemical methods (Markakis and Embs 1966), but their usage has been limited due to their negative effects on human health (Taylor and Bush 1986). Hence, it is imperative that the agents employed to manage enzymatic browning in foods or food products prioritize safety. Additionally, they should meet criteria of effectiveness and cost acceptability.
L-cysteine and ascorbic acid are the most widely used inhibitory agents approved by the EU commission regulation (Pizzocaro et al. 1993). Pizzocaro reported that ascorbic acid at 0.01–56.8 mM acts as an antioxidant, reducing o-quinone back to the original phenol compound, while L-cysteine at 40–100 mM makes it possible to form complexes with o-quinones (Doğru and Erat 2012; Siddiq and Dolan 2017). Inspired by this information, the inhibition potential of potato PPO enzyme of new quinazoline derivatives was investigated in our study.
To examine the PPO inhibition potentials of the synthesized quinazoline derivatives, the enzyme from potato, which is widely used in PPO studies, was purified by an affinity column. The inhibition potentials of quinazoline derivatives on the pure enzyme were determined by calculating IC50 values and Ki constants. The IC50 values for molecules 2–7 were calculated as 0.085, 1.145, 0.106, 6.86, 0.52, and 7.07 µM, respectively. Li et al. reported that the IC50 values of ascorbic acid, L-cysteine and reduced glutathione for potato polyphenol oxidase were 0.352, 0.370, 0.487 mM, respectively (Li 2020). Considering these IC50 values, it can be seen that the quinazoline derivative compounds in our study show better inhibition than known PPO inhibitors.
To further investigate the inhibitory type of quinazoline derivatives on PPO during the oxidation of catechol, the Lineweaver–Burk plots were used. Ki constants, which are inhibitory-enzyme binding constants, were calculated by using Lineweaver–Burk graphs (Fig. 2) as 0.16 ± 0.0620, 0.906 ± 0.3029, 0.055 ± 0.0171, 9.363 ± 2.5809, 0.773 ± 0.3204, 7.863 ± 1.9107 µM, respectively. In addition, Lineweaver–Burk plots were also used to evaluate the inhibition type. All the quinazoline derivatives were competitive inhibitors of PPO and hindered the enzymatic reaction by competing with the substrate for the active site of free PPO molecules. Inhibition parameters were summarized in Table 1. Previous studies showed that arabinose caused a mixed-type inhibition on PPO activity and Ki was 1.10 mM (Hu et al. 2012). Chlorogenic acid inhibited PPO activity in a mixed-type manner and Ki was 0.3577 mM (Cheng et al. 2020). In addition, isophthalic acid could lead to a mixed-type inhibition on PPO activity and Ki was 17.8 mM (Si et al. 2011). Compared with these compounds, the Ki of PPO activity in quinazoline derivatives was lower than that in chlorogenic acid, arabinose or isophthalic acid treatment.

Lineweaver–Burk graphs of PPO with compounds 2–7
In silico studies
Docking analyse
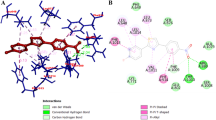
In order to clarify the possible binding poses and the affinity scores of the compounds 2–7 to PPO, docking studies were conducted and the interactions of the compounds at the active site of the enzyme were analysed. The 2D and 3D the best binding pose of the compound 2 on active sites of enzymes are presented in Fig. 3. The binding energy of all compounds calculated with Autodock and Vina is summarized in Table 1.

2D and 3D interaction patterns of compound 2 in active site of PPO
The results in Table 1 showed that docking affinities (kcal/mol) of compound 2–7 at Autodock 4.2 varied from − 6.95 to 7.62 and compounds scores varied from 7.6 to 8.0 at Autodock Vina. In in vitro inhibition results, compound 2 had the highest inhibitory efficiency with the IC50 value of 0.085 µM.
According to the best-fitting pose, compound 2 had a hydrogen bond with Asn 112 and Asn. It also interacted with Leu 447 and Phe 270 with π-sigma interactions and showed π-π and π-cation interaction with His 111 and Phe 270, respectively. Furthermore, compound 2 exhibited weak interactions with His 241, Gly 268, Ser 242, Ser 246, Glu 231, Gln 109, Val 110 and Arg 415. The gatekeeper residue Phe 270 stabilizes substrates via π interactions and this residue plays an important role during substrate binding (Kampatsikas et al. 2019a, b). All the molecules synthesized in our study interact with Phe270 and are also in balance with at least 2 hydrogen bonds in site, as supported by in vitro results, they are thought to compete with substrates for this region.
MD simulation
MD simulations are reasonable role on the screening the atoms movement, clarifying the structural fluctuations, flexibility, stability, and mainly protein–inhibitor interactions, RMSD and RMSF yields identifying the potential active compounds.
At the end of the simulation process, the dynamic motion of compound 2—PPO complex was monitored RMSD, RMSF, Rg, number of hydrogen bonds.
The comparison between the stability of the enzyme-inhibitor complex and enzyme was assessed using the backbone RMSD. The RMSD value of PPO enzyme stabilized after 3 ns in the average value of (0.25 nm), increased to a plateau of 0.3 nm at 65th ns to 70th ns, then back to 0.25 nm and to the end of simulation. The compound-backbone RMSD was a convenient measure of stability of interaction. In our simulation compound-backbone, RMSD analysis revealed that fluctuations minimized after 5 ns to 35 ns within the range of 0.25–0.4 nm, then a slight decreasing trend to 70 ns in the range of 0.2–0.3 nm. Between 70 and 160 ns, the former fluctuation trend repeated (0.25–0.4 nm). From 160 to 200 ns, a wider range of fluctuations (0.25 to 0.5 nm) were observed. These observations suggest that the complex structure of inhibitor and protein was reasonably stable, and the complex reaches dynamic equilibrium. RMS fluctuations of PPO were highest in residues from 80 to 120, and 250 to 270, indicating that ligand interacts mostly active side of PPO, as other residues present narrow fluctuations one can assume safely that interaction of ligand was not unstabilize PPO during simulation. Nonetheless, 0.7 nm fluctuations in residues 250 to 270 might be explaining the high number of hydrogen bond formation/deformations in the system. In Fig. 4, hydrogen bond formation throughout simulation was presented. According to graph, at least eighty per cent of the simulation time, three hbond were observed, which may account for the high affinity estimated in the in vitro experiment. High value of Rg indicates the expansion in the structural conformation of proteins, as low value is specifying the greater compactness and limit the system volume availability (Gupta et al. 2020). PPO is relatively a big protein containing 506 amino acids. Its gyration radius had fluctuated between 2.2 and 2.9 nm in our simulation. This indicates a significant stability, even after ligand interaction. Therefore, we can safely assume compound 2-PPO complex remained stable throughout the equilibrated phase of the simulation.

RMSD of PPO and PPO ligand complex (A), H bonds number formed during simulation (B), RMSF of PPO (C) and Rg of PPO (D)
DFT studies
The affinity of chemical compounds to proteins assigns their pharmacological values. These values especially rely on the electronic structure of the compounds. Density functional theory (DFT) elucidates values such as the electronic structure of the molecule and intermolecular charge transfer. These values like the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of the compounds provide data about kinetic stability, shape, conformations, and reactivity of the relevant compounds (Fig. 5) (Franke 1984; Zhou and Parr 1990; Aihara 1999).

DFT optimized structure of compound 2 and compound 4, visualization of LUMO and HOMO energy levels
For optimization and other DFT calculations of compound 2–7, molecules were subjected to density functional theory under the B3LYP model /6-311G (d, p) basis set. The optimized structure of compounds 2 and 4 which is the having the best PPO inhibiting activity is shown in Fig. 4. Additionally, the HOMO and LUMO energies of the synthesized compounds were calculated and these values were used to determine the global reactivity descriptors, namely, ionization potential (EHOMO), electron affinity ELUMO, chemical potential, hardness, electrophilicity index (Koopmans 1934, Parr, Szentpály et al. 1999) (Table 2). According to Table 2, the HOMO/LUMO energy gap value is highest at compound 2, on the contrary, compound 3 has a minimum with the value of 3.777 eV and this value account for the easy electron transfer capacity. The ionization potential value of compound 5 is slightly more than other compounds when the hardness of compound 2 is highest. According to IP value, compound 6 is expected to be more stable/less reactive and weak inhibitor, but in the light of the data obtained from in vitro and in silico studies, it was clearly observed that compound 2 has the highest inhibition values. Therefore, this situation was explained that the bonded groups to core structure effects the inhibition value rather than the electronic structure. Especially, the number of attached bromine atoms dramatically reduces the inhibition values (see compound 6).
Conclusion
In this research paper, we designed and synthesized a series of quinazoline derivatives having new phenolic moieties. All the synthesized compounds 2–7 were evaluated for their in vitro and in silico polyphenol oxidase catalyse capacity limiting effect. Compound 2 in the active site interacted with the Phe 270, key residue for activity, and had formation of at least two H bond with protein. Similarly, MD simulation showed that compound 2 formed stable complex with PPO throughout 200 ns. These data make compound 2 that is a strong potential candidate for PPO inhibition. Additionally, we thought that new quinazoline derivatives, which have the potential to be used as anti-browning agents in food processing, will contribute to the food industry.
References
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Ahmad I (2017) An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. MedChemComm 8(5):871–885
Ahmed MF, Youns M (2013) Synthesis and biological evaluation of a novel series of 6, 8-D ibromo-4 (3 H) quinazolinone derivatives as anticancer agents. Arch Pharm 346(8):610–617
Aihara J-I (1999) Reduced HOMO−LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J Phys Chem A 103(37):7487–7495
Aksoy M (2020) A new insight into purification of polyphenol oxidase and inhibition effect of curcumin and quercetin on potato polyphenol oxidase. Protein Expr Purif 171:105612
Alagarsamy V, Raja Solomon V, Sheorey R, Jayakumar R (2009) 3-(3-Ethylphenyl)-2-substituted hydrazino-3H-quinazolin-4-one derivatives: new class of analgesic and anti-inflammatory agents. Chem Biol Drug Des 73(4):471–479
Arslan O, Erzengin M, Sinan S, Ozensoy O (2004) Purification of mulberry (Morus alba L.) polyphenol oxidase by affinity chromatography and investigation of its kinetic and electrophoretic properties. Food Chem 88(3):479–484
Biovia DS, DSME R (2017) San Diego: Dassault Systèmes, 2016.
Bottino A, Degl’Innocenti E, Guidi L, Graziani G, Fogliano V (2009) Bioactive compounds during storage of fresh-cut spinach: the role of endogenous ascorbic acid in the improvement of product quality. J Agric Food Chem 57(7):2925–2931
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J chem phys 126(1):014101. https://doi.org/10.1063/1.2408420
Chazarra S, Cabanes J, Escribano J, Garcia-Carmona F (1996) Partial purification and characterization of latent polyphenol oxidase in iceberg lettuce (Lactuca sativa L.). J Agric Food Chem 44(4):984–988
Cheng D, Wang G, Tang J, Yao C, Li P, Song Q, Wang C (2020) Inhibitory effect of chlorogenic acid on polyphenol oxidase and browning of fresh-cut potatoes. Postharvest Biol Technol 168:111282
Contente ML, Annunziata F, Cannazza P, Donzella S, Pinna C, Romano D, Tamborini L, Barbosa FG, Molinari F, Pinto A (2021) Biocatalytic approaches for an efficient and sustainable preparation of polyphenols and their derivatives. J Agric Food Chem 69(46):13669–13681
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. Springer, Chemical biology, pp 243–250
DeLano WL (2002) Pymol an open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr 40(1):82–92
Doğru YZ, Erat M (2012) Investigation of some kinetic properties of polyphenol oxidase from parsley (Petroselinum crispum, Apiaceae). Food Res Int 49(1):411–415
eddine Derardja A, Pretzler M, Kampatsikas I, Barkat M, Rompel A (2019) Inhibition of apricot polyphenol oxidase by combinations of plant proteases and ascorbic acid. Food Chemistry: X 4:100053
Flurkey WH (1986) Polyphenoloxidase in higher plants: immunological detection and analysis of in vitro translation products. Plant Physiol 81(2):614–618
Fortea M, López-Miranda S, Serrano-Martínez A, Carreño J, Núñez-Delicado E (2009) Kinetic characterisation and thermal inactivation study of polyphenol oxidase and peroxidase from table grape (Crimson Seedless). Food Chem 113(4):1008–1014
Franke R (1984) Theoretical drug design methods. De Gruyter, Germany
Georgey H, Abdel-Gawad N, Abbas S (2008) Synthesis and anticonvulsant activity of some quinazolin-4-(3 H)-one derivatives. Molecules 13(10):2557–2569
Gouzi H, Depagne C, Coradin T (2012) Kinetics and thermodynamics of the thermal inactivation of polyphenol oxidase in an aqueous extract from Agaricus bisporus. J Agric Food Chem 60(1):500–506
Guiamba IR, Svanberg U (2016) Effects of blanching, acidification, or addition of EDTA on vitamin C and β-carotene stability during mango purée preparation. Food Sci Nutr 4(5):706–715
Gupta PSS, Bhat HR, Biswal S, Rana MK (2020) Computer-aided discovery of bis-indole derivatives as multi-target drugs against cancer and bacterial infections: DFT, docking, virtual screening, and molecular dynamics studies. J Mol Liq 320:114375
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Chem 4(1):1–17
Hricovíniová Z, Hricovíni M, Kozics K (2018) New series of quinazolinone derived Schiff’s bases: synthesis, spectroscopic properties and evaluation of their antioxidant and cytotoxic activity. Chem Pap 72(4):1041–1053
Hu W-J, Yan L, Park D, Jeong HO, Chung HY, Yang J-M, Ye ZM, Qian G-Y (2012) Kinetic, structural and molecular docking studies on the inhibition of tyrosinase induced by arabinose. Int J Biol Macromol 50(3):694–700
Ismail MA, Barker S, Abou El Ella DA, Abouzid KA, Toubar RA, Todd MH (2006) Design and synthesis of new tetrazolyl-and carboxy-biphenylylmethyl-quinazolin-4-one derivatives as angiotensin II AT1 receptor antagonists. J Med Chem 49(5):1526–1535
Janovitz-Klapp AH, Richard FC, Goupy PM, Nicolas JJ (1990) Inhibition studies on apple polyphenol oxidase. J Agric Food Chem 38(4):926–931
Kampatsikas I, Bijelic A, Rompel A (2019a) Biochemical and structural characterization of tomato polyphenol oxidases provide novel insights into their substrate specificity. Sci Rep 9(1):4022
Kampatsikas I, Bijelic A, Rompel A (2019b) Biochemical and structural characterization of tomato polyphenol oxidases provide novel insights into their substrate specificity. Sci Rep 9(1):1–13
Khan I, Zaib S, Batool S, Abbas N, Ashraf Z, Iqbal J, Saeed A (2016) Quinazolines and quinazolinones as ubiquitous structural fragments in medicinal chemistry: an update on the development of synthetic methods and pharmacological diversification. Bioorg Med Chem 24(11):2361–2381
Khurana S, Venkataraman K, Hollingsworth A, Piche M, Tai T (2013) Polyphenols: benefits to the cardiovascular system in health and in aging. Nutrients 5(10):3779–3827
Knizia G (2013) Intrinsic atomic orbitals: an unbiased bridge between quantum theory and chemical concepts. J Chem Theory Comput 9(11):4834–4843
Koopmans T (1934) Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica 1(1–6):104–113
Krishnan S, Ganguly S, Veerasamy R, Jan B (2011) Synthesis, antiviral and cytotoxic investigation of 2-phenyl-3-substituted quinazolin-4 (3H)-ones. Eur Rev Med Pharmacol Sci 15(6):673–681
Kuijpers TF, Narváez-Cuenca C-E, Vincken J-P, Verloop AJ, van Berkel WJ, Gruppen H (2012) Inhibition of enzymatic browning of chlorogenic acid by sulfur-containing compounds. J Agric Food Chem 60(13):3507–3514
Kuijpers TF, Gruppen H, Sforza S, van Berkel WJ, Vincken JP (2013) The antibrowning agent sulfite inactivates Agaricus bisporus tyrosinase through covalent modification of the copper-B site. FEBS J 280(23):6184–6195
Kumar P, Tomar V, Joshi RK, Nemiwal M (2022) Nanocatalyzed synthetic approach for quinazoline and quinazolinone derivatives: a review (2015-present). Synth Commun 52(6):795–826
Li F (2020) Purification, kinetic parameters, and isoforms of polyphenol oxidase from “Xushu 22” sweet potato skin. J Food Biochem 44(11):e13452
Li AN, Li S, Zhang YJ, Xu XR, Chen YM, Li HB (2014) Resources and biological activities of natural polyphenols. Nutrients 6(12):6020–6047
Liu W, Zou L-Q, Liu J-P, Zhang Z-Q, Liu C-M, Liang R-H (2013) The effect of citric acid on the activity, thermodynamics and conformation of mushroom polyphenoloxidase. Food Chem 140(1–2):289–295
Manasa K, Sidhaye R, Radhika G, Nalini C (2011) Synthesis, antioxidant and anticancer activity of quinazoline derivatives. J Curr Pharma Res 1(2):101
Markakis P, Embs R (1966) Effect of sulfite and ascorbic acid on mushroom phenol oxidase. J Food Sci 31(6):807–811
Mathew A, Parpia H (1971) Food browning as a polyphenol reaction. In: Chichester CO, Mrak EM, Stewart GF (eds) Advances in food research. Elsevier, pp 75–145
Mayer AM (2006) Polyphenol oxidases in plants and fungi: going places? A review. Phytochemistry 67(21):2318–2331
McEvily AJ, Iyengar R, Otwell WS (1992) Inhibition of enzymatic browning in foods and beverages. Crit Rev Food Sci Nutr 32(3):253–273
Neese F (2022) Software update: the ORCA program system—Version 5.0. Wiley Int Rev: Comput Mol Sci 12(5):e1606
Nerkar A, Saxena A, Ghone S, Thaker A (2009) In silico screening, synthesis and in vitro evaluation of some quinazolinone and pyridine derivatives as dihydrofolate reductase inhibitors for anticancer activity. E-J Chem 6(S1):S97–S102
Nirmal NP, Benjakul S (2009) Effect of ferulic acid on inhibition of polyphenoloxidase and quality changes of Pacific white shrimp (Litopenaeus vannamei) during iced storage. Food Chem 116(1):323–331
Öztürk C, Aksoy M, Küfrevioğlu Öİ (2020) Purification of tea leaf (Camellia sinensis) polyphenol oxidase by using affinity chromatography and investigation of its kinetic properties. J Food Measurement Charact 14(1):31–38
Panneerselvam P, Rather BA, Reddy DRS, Kumar NR (2009) Synthesis and anti-microbial screening of some Schiff bases of 3-amino-6,8-dibromo-2-phenylquinazolin-4(3H)-ones. Eur J Med Chem 44(5):2328–2333
Parr RG, Szentpály Lv, Liu S (1999) Electrophilicity index. J Am Chem Soc 121(9):1922–1924
Patel NB, Patel VN, Patel HR, Shaikh FM, Patel JC (2010) Synthesis and microbial studies of (4-oxo-thiazolidinyl) sulfonamides bearing quinazolin-4 (3H) ones. Acta Pol Pharm 67:267–275
Pati B, Banerjee S (2013) Quinazolines: an illustrated review. J Adv Pharm Edu Res 3(3):136–151
Pizzocaro F, Torreggiani D, Gilardi G (1993) Inhibition of apple polyphenoloxidase (PPO) by ascorbic acid, citric acid and sodium chloride. J Food Process Preserv 17(1):21–30
Queiroz C, Lopes MLM, Fialho E, Valente-Mesquita VL (2008) Polyphenol oxidase: characteristics and mechanisms of browning control. Food Rev Intl 24(4):361–375
Raghavendra NM, Thampi P, Gurubasavarajaswamy PM, Sriram D (2007) Synthesis and antimicrobial activities of some novel substituted 2-imidazolyl-N-(4-oxo-quinazolin-3(4H)-yl)-acetamides. Chem Pharm Bull 55(11):1615–1619
Sak K (2012) Chemotherapy and dietary phytochemical agents. Chemother Res Pract 2012:1–11
Saravanan G, Pannerselvam P, Prakash CR (2010) Synthesis and anti-microbial screening of novel schiff bases of 3-amino-2-methyl quinazolin 4-(3H)-one. J Adv Pharm Technol Res 1(3):320
Scalbert A, Manach C, Morand C, Rémésy C, Jiménez L (2005) Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr 45(4):287–306
Si Y-X, Yin S-J, Park D, Chung HY, Yan L, Lü Z-R, Zhou H-M, Yang J-M, Qian G-Y, Park Y-D (2011) Tyrosinase inhibition by isophthalic acid: kinetics and computational simulation. Int J Biol Macromol 48(4):700–704
Siddiq M, Dolan K (2017) Characterization of polyphenol oxidase from blueberry (Vaccinium corymbosum L.). Food Chem 218:216–220
Smits RA, Adami M, Istyastono EP, Zuiderveld OP, van Dam CM, de Kanter FJ, Jongejan A, Coruzzi G, Leurs R, de Esch IJ (2010) Synthesis and QSAR of quinazoline sulfonamides as highly potent human histamine H4 receptor inverse agonists. J Med Chem 53(6):2390–2400
Taylor S, Bush R (1986) Sulfites as a food ingredients. Food Technol (Chicago) 40(6):47–52
Tinello F, Lante A (2018) Recent advances in controlling polyphenol oxidase activity of fruit and vegetable products. Innov Food Sci Emerg Technol 50:73–83
Tokalı FS, Taslimi P, Demircioğlu İH, Karaman M, Gültekin MS, Şendil K, Gülçin İ (2021) Design, synthesis, molecular docking, and some metabolic enzyme inhibition properties of novel quinazolinone derivatives. Arch Pharm 354(5):2000455
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Verhaeghe P, Azas N, Gasquet M, Hutter S, Ducros C, Laget M, Rault S, Rathelot P, Vanelle P (2008) Synthesis and antiplasmodial activity of new 4-aryl-2-trichloromethylquinazolines. Bioorg Med Chem Lett 18(1):396–401
Zhou Z, Parr RG (1990) Activation hardness: new index for describing the orientation of electrophilic aromatic substitution. J Am Chem Soc 112(15):5720–5724
Zhou L, Liu W, Xiong Z, Zou L, Chen J, Liu J, Zhong J (2016) Different modes of inhibition for organic acids on polyphenoloxidase. Food Chem 199:439–446
Zhou L, Liao T, Liu W, Zou L, Liu C, Terefe NS (2020) Inhibitory effects of organic acids on polyphenol oxidase: from model systems to food systems. Crit Rev Food Sci Nutr 60(21):3594–3621
Zhu L, Zhu L, Murtaza A, Liu Y, Liu S, Li J, Iqbal A, Xu X, Pan S, Hu W (2019) Ultrasonic processing induced activity and structural changes of polyphenol oxidase in orange (Citrus sinensis Osbeck). Molecules 24(10):1922
Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32(11):2359–2368
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Öztürk, C., Tokali, F.S., Öztekin, A. et al. Synthesis of quinazoline derivatives with new phenolic moieties: in vitro and in silico evaluations as alternative polyphenol oxidase inhibitors. Chem. Pap. (2024). https://doi.org/10.1007/s11696-024-03670-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11696-024-03670-5