
Abstract
Neurodegenerative diseases are characterized by the presence of amyloid deposition. Thioflavin T (ThT) has been one of the molecules of choice to attempt the detection of the amyloid deposits, but ThT is unable to cross blood–brain barrier, due to its low lipophilicity. Therefore, there is strong motivation to design and develop new compounds for in vitro fibril detection as well as for in vivo amyloid imaging. Additionally, the importance and critical role of oxidative stress in the onset/progression of some neurodegenerative disorders, and therefore, the efficacy of aurone compounds in inhibiting the resulting toxicity have been frequently reported. In this study, we report the synthesis of some benzofuranone compounds and examine their antioxidant inhibitory property. Furthermore, to establish the potential detection of synthesized compounds to amyloid aggregates, their in vitro binding to some non-disease related amyloidogenic proteins were characterized. Analyses of the in vitro binding studies showed that compounds 3 and 4 bind to the fibril structures successfully while compounds 1, 2 and 5 indicated a low affinity binding to amyloid. Additionally, compounds 3 and 4 exhibited very good antioxidant properties. Furthermore, these compounds have a great potential as fluorescent probes for detecting amyloid aggregation for further investigations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyloid fibrils are insoluble fibrillary protein, possess a cross β-sheet quaternary structure which formed by normally soluble proteins (or protein fragments) [1]. Amyloid fibrils have been associated with more than 50 human diseases [2], known as amyloidosis, a group of diseases requiring the self-aggregation of specific proteins (or protein fragments) into cytotoxic filamentous deposits [3]. Amyloidosis, including Alzheimer’s disease (AD), Parkinson’s disease, systemic amyloidosis, and type-2 diabetes are pathologically characterized by the presence of amyloid fibrils which are built up in one or more diseased organs [3]. For example, AD is a neurodegenerative disorder widely abundant in elderly people. Pathological hallmarks of AD include intracellular neurofibrillary tangles consisting of insoluble deposits of hyperphosphorylated microtubule-associated tau protein and extracellular amyloid plaques deposition, mainly composed by neurotoxic Aβ peptides [4]. Despite the many pathological characteristics of AD, one of the most consensual hypothesis that explains the disease process is the amyloid hypothesis. It states that the gradual accumulation and aggregation of the hydrophobic Aβ peptides can directly or indirectly result in progressive synaptic and neurotic injury through the generation of a complex cascade of molecular events, which subsequently lead to hyperphosphorylation of tau and formation of neurofibrillary tangles [5]. Indeed, Aβ accumulation, oligomerization and deposition within the brain are the hallmarks of AD pathogenesis, contributing to the imbalances observed in Aβ production and clearance in AD [6].
On the other hand, during the process of aggregation, Aβ generates hydrogen peroxide, a mitochondrial redox product that requires oxygen and greatly potentiated by Fe2+ and Cu+ [7, 8], leading to oxidative damage of various cellular and molecular components, such as proteins, lipids and nucleic acids, and it occurs by reactive oxygen species (ROS). Several studies report directly demonstrated extensive oxidative damage in association with AD (oxidative-stress-hypothesis) [7, 9, 10]. Brains of AD patients exhibit high amounts of oxidized macromolecules as well as impaired cellular function and cell death [11, 12]. Several studies also showed oxidative stress induced by Aβ can be inhibited by antioxidants [13,14,15]. Recent studies are focusing on the design and development of drugs that will protect or delay the progression of the disease, giving opportunities for alternative methods of prevention/treatment [16].
Detecting deposits of Aβ or Tau fibrils in the brain as well as design of tracers with specificity and improved binding affinity are of paramount importance for an early diagnosis of Alzheimer’s disease. Developing new molecules to be used as a marker of β-amyloid or Tau deposits in AD has been a goal of researchers for many years [17]. Understanding of molecular details of amyloidosis is required for developing new strategies to detect and control amyloid aggregation/formation and consequently, neurodegenerative disorders. Thioflavin T is a highly sensitive fluorescent marker of amyloid fibrils that has been widely used for in vitro biomedical assays. However, neither its complex photophysical behavior nor its binding mode to amyloid fibrils is still well understood. On the other hand, the same ThT-BSA/ThT- Aβ42 binding affinity as well as a similar huge fluorescence enhancement (that follows a typical complexation profile) both for bovine serum albumin (BSA, an all-α protein) and for Aβ42 has been documented [18]. This non-specific increase in fluorescence is attributed to the binding of ThT to the peptides and a resulting steric hindrance for the dye to undergo the twisting process. Additionally, ThT has a positive charge, and it seems that compounds without a permanent positive charge are supposed to be capable of crossing the blood–brain barrier (BBB) [19]. So, in this study, we synthesized and employed benzofuranone compounds (including aurone derivatives), as (1) fluorescent probes to quantitatively determine the amyloid fibrils made up of β-lactoglobulin (β-Lg), and (2) antioxidant compounds. The resulting data may be useful in providing mechanistic insights to develop potential diagnostic, curative, and/or preventive strategies in vivo against amyloid-related neurodegenerative disorders.
Materials and methods
Chemicals and reagents
Ammonium persulfate, bromophenol blue, glycerol, glycine, methylene bisacrylamide, SDS (sodium dodecyl sulphate), TEMED (N,N,N′,N′-tetramethylethylenediamine), HCl, ethanol, 3-methyle-2-benzothiazolinone hydrazine, cysteine, EDTA (ethylene diamine tetra-acetic acid), and ammonium sulfate were purchased from Merck (Darmstadt, Germany). Na2HPO4, KH2PO4, Tris, NaCl, NaOH, Na2CO3 were purchased from Applichem (Darmstadt, Germany). Bovine serum albumin was purchased from Sigma-Aldrich (St. Louis, MO). DEAE-sepharose resin was purchased from Pharmacia Biotech Inc. Acrylamide was purchased from Fluka (Sydney, Australia). All other chemicals were of the highest analytical grade of purity available and were used as obtained from suppliers. All solutions were prepared with double distilled water. Unless otherwise stated, all solutions were made in 20 mM sodium phosphate buffer (pH 7–7.4). A stock solution of ThT (10 mM) was freshly prepared in buffer and stored in the dark at 4 °C. The structure of compounds was characterized by IR, 1H-NMR spectra and mass spectrometry (MS). IR spectrum was recorded on a Shimadzu 470 spectrophotometer (KBrdisk). 1H-NMR spectrum was recorded on a Bruker FT-250 NMR spectrophotometer using DMSO-d6 as solvent and tetramethylsilane (TMS) as an internal standard. The MS analysis was performed using a MS system comprising a Finnegan MAT Spectra System P4000 pump coupled with a UV6000LP diode array detector and a Finnigan AQA mass spectrometer. A Cary-100 Bio (VARIAN) spectrophotometer was used for protein determination. All fluorescence measurements were performed in the ratio mode using a 1 cm cell in a Cary Eclipse (VARIAN) fluorescence spectrophotometer, equipped with a 150 W xenon lamp and a thermostatic cell holder, at room temperature or as stated. Appropriate vehicle controls were run in all experiments. All the reported results are averages of 2–3 separate experiments whenever the coefficients of variation were less than 5%.
Chemistry
Solution of 6-hydroxy-3-benzofuranones (1 mmol), and appropriate aryl aldehyde (1 mmol) in ethanol (5 ml) were added to piperazine (0.5 mmol) as the basic catalyst. The reaction mixture was heated in 80 °C for 12–24 h, then was allowed to stand at room temperature for the next 24 h. The precipitate was filtered, dried, and crystallized from acetic acid to afford pure 6-hydroxy-2-benzylidene-3-benzofuranones. Structural assignments of the products are based on their IR, 1H-NMR, MS and melting point [20, 21].
6-Hydroxy-2-(4ʹ-nitrobenzylidene)benzofuran-3(2H)-one (compound 1)
Yield = 74%; Orange solid; m.p. over 300 °C; 1H-NMR (250 MHz, DMSO-d6): δ 4.67 (s, OH aromatic), 6.67–6.72 (d, J = 8 Hz CH benzene), 6.79 (s, CH ethylene), 6.81 (s, CH benzene), 7.08–7.12 (d, J = 12 Hz, CH benzene), 7.18–7.22 (d, J = 8 Hz, CH benzene), 7.86–7.90 (d, J = 8 Hz, CH benzene) ppm; IR (KBr, cm−1): υ3326, 1763, 1723, 1500, 1385, 1280, 1153, 1058, 725; MS (m/z, %): 284 (M+, 16.5), 283 (100), 152 (33), 137 (55), 85 (70).
6-Hydroxy-2-(2ʹ-nitrobenzylidene) benzofuran-3(2H)-one (compound 2)
Yield = 63%; Orange solid; m.p. over 300 °C; 1H-NMR (250 MHz, DMSO-d6): δ 4.67 (s, OH aromatic), 6.72 (s, CH benzene), 6.94 (s, CH ethylene), 7.57–7.65 (m, CH benzene), 7.68–7.80 (m, CH benzene), 7.86–7.90 (d, J = 8 Hz, CH benzene), 7.88–7.92 (d, J = 8 Hz, CH benzene) ppm; IR (KBr, cm−1): υ3335, 1760, 1712, 1614, 1565, 1406, 1374, 1280, 1153, 1064, 727; MS (m/z, %): 284 (M+, 14), 283 (100), 152 (33), 137 (55), 85 (70).
6-Hydroxy-2-(2ʹ,3ʹ,4ʹ-trihydroxybenzylidene)benzofuran-3(2H)-one (compound 3)
Yield = 57%; Orange solid; m.p. over 300 °C; 1H-NMR (250 MHz, DMSO-d6): δ 6.43 (s, OH aromatic), 6.64–6.67 (d, J = 8 Hz CH benzene), 6.72 (s, CH benzene), 6.75 (s, CH benzene), 6.79 (s, CH ethylene), 7.07–7.12 (d, J = 12 Hz, CH benzene), 7.86–7.90 (d, J = 8 Hz, CH benzene) ppm; IR (KBr, cm−1): υ3372, 1752, 1712, 1608, 1500, 1406, 1220, 1153, 1059, 735; MS (m/z, %): 287 (M+, 12.5), 286 (100), 176 (30), 126 (30), 110 (50), 85 (90).
6-hydroxy-2-(3,4,5-trihydroxybenzylidene)benzofuran-3(2H)-one (compound 4)
Yield = 61%; Orange solid; m.p. over 300 °C; 1H-NMR (250 MHz, DMSO-d6): δ 6.21 (s, OH aromatic), 6.68 (s, CH ethylene), 6.72 (s, CH benzene), 6.85 (s, CH benzene), 7.07–7.12 (d, J = 8 Hz, CH benzene), 7.86–7.90 (d, J = 8 Hz, CH benzene) ppm; IR (KBr, cm−1): υ3351, 1761, 1715, 1602, 1500, 1406, 1220, 1153, 1066, 725; MS (m/z, %): 287 (M+, 10), 286 (100), 176 (30), 126 (30), 110 (50), 85 (90).
6-Hydroxy-2-(4′-hydroxy-3′-methoxybenzylidene)benzofuran-3(2H)-one (compound 5)
Yield 80%; m.p. = 280–282 °C; 1H-NMR (300 MHz, DMSO-d6): δ 7.61 (d, J = 8.7 Hz, 1H,H5‚), 7.49 (d, J = 8.7 Hz, 1H, H6), 7.53 (s, 1H, H2‚), 6.91 (d, J = 8.8 Hz, 1H, H4), 6.80(s, 1H, H7), 6.71 (d, J = 8.7 Hz, 1H, H5), 6.80 (s, 1H, =CH), 3.85 (s, 3H, OCH3), 3.80 (s, 2H, OH) ppm; 13C NMR (DMSO-d6, 75 MHz): δ182.3, 167.5, 165.1, 149.2, 147.4, 146.7, 128.1, 125.6, 121.5, 116.4, 115.3, 111.6, 101.2, 56.2 ppm; IR (KBr, cm−1): υ 1743 (C=O), 3250 (OH).
Protein purification/determination
β-Lg was purified according to the method described by Ghadami et al. [22]. The purity of protein was checked by one-dimensional 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in vertical slab gel electrophoresis chamber [23]. Protein concentrations of purified β-Lg fractions were measured using Compassion Blue dye reagent, measured at 595 nm, according to the Bradford method with bovine serum albumin (BSA) as a protein standard [24].
Sample preparation
Preparation of amyloid aggregate
Amyloid aggregate made by β-Lg was monitored using fluorescent dyes [25, 26]. The β-Lg was converted to amyloid-like fibrils by the following protocol; 20–30 mg of β-Lg was dissolved in 2–3 ml phosphate buffer 10 mM at pH 2. The protein solution was incubated about 400 min at 80 °C. The amyloid formation procedure was monitored by ThT probe every single 12 h [22] and finally the fibril was determined by transmission electron microscopy (TEM).
Preparation of amorphous aggregate
Amorphous aggregate was composed of bovine serum albumin (BSA) by the following method; 8 mg of BSA protein was dissolved in sodium nitrate buffer 50 mM at pH 5–5.5 [27]. For obtaining the modify structure of protein, the protein solution was incubated about 25–30 min at 80–90 °C. The transparent soluble protein getting turbid when the native structure of protein became denature visibly and gradually [28] and was confirmed by atomic force microscopy (AFM) imaging.
Preparation of stock solutions of compounds and BSA/β-Lg
10 mM dye stock solutions were prepared by dissolving the dye in DMSO [21, 26]. The concentrations of proteins in stock solutions were 1 mg/ml for β-Lg (both native and amyloid aggregates). The concentration of stock BSA proteins were 2 mg/ml (both native and amorphous aggregates). Working solutions of free dyes at 10 mM were prepared by dilution of the dye stock solution in 20 mM sodium phosphate buffer (pH 7.4). Dye-protein complexes solutions were prepared by mixing an aliquot of the dye stock solution (2.0 µL) with an aliquot (1, 2, and 3 µl) of native or aggregate proteins in 20 mM sodium phosphate buffer. Concentration of the proteins in working solution were 0, 0.05, 0.1, 0.15, and 0.2 mg/ml, for native, amyloid and amorphous aggregates, separately. All working solutions were prepared immediately before the experiment.
AFM and TEM imaging
The AFM imaging of amorphous aggregate of Bovine serum albumin (BSA) was performed in dynamic force operating mode and noncontact Mounted cantilever (Nanosurf mobile S. Switzerland) using high frequency (170 kHz) by silicon cantilevers with thickness 7 mm, length 225 mm, and width 38 mm. Images were treated using the software installation instructions for Nano surf Mobile S version 1.8.
Samples of fibrillar dispersions of β-Lg were prepared for TEM using negative staining. A droplet of the β-Lg sample (20 mg/ml), which had been diluted 1:10 in water, was deposited onto carbon support film on a 400-mesh copper grid. After 1 min excess liquid was removed, using filter paper. Then, a drop of staining solution (0.1% (w/w) phosphotungstic acid) was added, and excess solution was removed after 30 s using a filter paper. Electron micrographs were taken using a Philips EM208 field emission scanning electron microscope. Bright field TEM images were acquired at 80 kV [22].
Spectroscopic measurements
UV–visible spectra were evaluated on a Carry-100 Bio UV–visible spectrophotometer (Varian, Australia). To determine the appropriate wavelengths of fluorescence studies, including excitation and emission, the UV–Vis spectroscopy was used. Spectrum of each dye was collected between 200 and 600 nm for choosing the best λmax to be used as an excitation wavelength of fluorescence spectroscopy (data not shown). Fluorescence spectra were collected on a Cary Eclipse fluorescence spectrophotometer (Varian, Australia). Fluorescence spectra were measured with excitation and emission slit widths of 10 nm. The study of the fluorescent spectra of each dye in the presence of native and aggregated form of both β-Lg [29] and BSA proteins were performed, separately. In vitro binding studies were conducted at least in triplicate by the procedures described in detail by Groenning and his co-workers [30].
Docking studies
Molecular docking was performed using the Auto Dock 4.2 software [31]. The structures for each compound were mapped using ChemDraw [32], and after preparing a 3-D coordinate file in Chem 3D (both in Chem Office package ver. 15), the energy minimization and final optimization of the 3-D structures were performed using the steepest descent algorithm of Avogadro software [33]. The coordinate file for the β-amyloid was provided by rcsb protein datacenter coded with 2beg. MGL tools package was used for preparation of coordinate files containing information on the charges, active torsions and atomic type of the Auto Dock force field. Then, Gasteiger charges were added to each atom, and all the torsions were considered active for all ligands [31]. The search space was chosen to accommodate all of the protein spaces available to the ligand. Preparation of energetic maps of ligand-related atom types in the search space was done using the Auto Grid 4 software [34]. Finally, the interaction structures with the lowest energy were obtained from Lamarck’s genetic algorithm [34] for 100 runs.
Antioxidant assays
DPPH radical scavenging assay
The phenolic compounds were dissolved in appropriate solvent mixed with 1 ml of 0.2 mM 2,2-diphenyl-1-picrylhydrazyl radical (DPPH) in ethanol, and final volume was adjusted to 2 ml. Mixtures were variously shaken and left for 30 min in the dark. Absorbance was measured at 517 nm using a UV–Vis spectrophotometer. 1 ml of 0.2 mM DPPH diluted in 1 ml of ethanol was used as control. Neutralization of DPPH radical was calculated using the equation: S (%) = 100 (Ao − As)/Ao, where Ao is the absorbance of the control (containing all reagents except the test compound) and As is the absorbance of the test sample. Results were compared to activity of Trolox [35]. The IC50 values represented the concentration of the test compounds that caused 50% inhibition are shown in Table 3.
Ferric reducing antioxidant power (FRAP) assay
The FRAP assay reagent was prepared by adding 10 vol of 30 mM acetate buffer, pH 3.6 (1 g sodium acetate and 16 ml glacial acetic acid), 1 vol of 10 mM 2,4,6-tripyridyl-triazine prepared in 40 mM HCl and 1 vol of 20 mM FeCl3. The mixture was diluted to 1/3 with methanol and pre-warmed at 37 °C. This reagent (3 ml) was mixed with 0.1 ml diluted the test compounds. The mixture was shaken and incubated at 37 °C for 8 min and the absorbance was read at 593 nm. A blank with only 0.1 ml methanol was used for calibration [36, 37]. The difference in absorbance between the tested sample and the blank reading was calculated and the data were expressed as mM of ferric reduced to ferrous form (Table 3).
Results and discussion
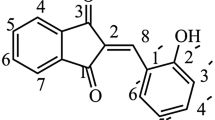
Pervious in vitro studies have suggested that polyphonolic compounds (PCs) from food products can be effective in targeting Aβ [16]. Among all of them, flavonoids have been of interest due to their anti-oxidant, anti-inflammation properties, targeting metal-Aβ40, modulate metal-Aβ40 aggregation in vitro and diminish cytotoxicity induced by metal-Aβ40 [38]. Recently, it was reported that flavonoids including flavone, and aurone serve as useful molecular probes in the development of imaging agents for β-amyloid plaques [39, 40]. To explore more useful candidates for amyloid imaging probes, we selected one of the flavonoids, aurone, as a new core structure [21, 41]. We synthesis 5 aurone derivatives (Fig. 1) as shown in Fig. 2.

Chemical structures of synthesized compounds 1–5. See also aurone structures for comparison

Synthesis of compounds 1–5: reagents and conditions
To characterize the binding assay, the fluorescent spectra of each dye in the presence of native and aggregated forms of two model proteins: β-Lg [29], native and amorphous BSA [42] were measured, separately. Native β-Lg have been found to be converted to amyloid fibrils under various experimental conditions [22, 43]. There are three types of aggregates were generated upon heating β-Lg at increasing pH from 2.0 to 5.8 to 7.0: rod-like aggregates, spherical aggregates, and worm-like primary aggregates, respectively [44, 45]. In this investigation, to drive purified β-Lg (Fig. 3a) toward amyloid fibril formation, the native solutions at pH 2.0, for 400 min was incubated in 80 °C [22]. Thereafter, formation of amyloid fibrils by native β-Lg was verified using ThT fluorescence spectroscopy as well as Congo red binding assay [46] (data not shown). Additionally, additional characterization of amyloid fibrils was performed using TEM (Fig. 3b). The amorphous form of BSA was obtained according the procedure described in material and methods section and as indicated in Fig. 3c, the amorphous aggregation of BSA was verified using the AFM.

a SDS-PAGE pattern of purified β-Lg (lane 2) and molecular size markers (lane 1) (purity 99.0%). b Negatively stained electron micrographs of aggregated β-Lg by TEM. Samples (20 mg/ml) were incubated at pH 2.0 (for native β-lg) and heated at 80 °C for specified time. Scale bars represent 100 nm. (c) Surface AFM micrographs in air of the dry protein (BSA) films adsorbed onto mica surfaces. Further details are given in Experimental procedures
Binding of compounds 1–5 to the β-Lg amyloid aggregate was examined by observing fluorescence emission spectra between 300 and 600 nm, using specific excitation wavelengths (410, 340, 420, 345, and 380 nm for compounds1–5, respectively) and at a fixed concentration (40 µM) of compounds, the excitation wavelength for ThT was set at 440 nm. As indicated in (Fig. 4) the fluorescence intensities (402, 478, 490, 392, and 505 nm for compounds 1–5, respectively) increased upon interaction with amyloid aggregates. Moreover, as the amount of amyloid aggregates (corresponding to the concentration of β-Lg solution) increased, the fluorescence intensities were enhanced proportionally, so that the maximum fluorescence emissions increased linearly as a function of amyloid concentration. Unlike the amyloid aggregates, however, the fluorescence intensity of the compounds (except compound 5) did not undergo a significant change upon interaction with the soluble (monomeric) form of β-Lg (Fig. 4).

Binding fluorescence spectra of compounds 1–5 in the present of native β-Lg (filled circle), native BSA (filled square), β-Lg amyloid aggregates (open circle) and BSA amorphous aggregates (open square). Compounds 1–5 binding fluorescence spectra between 300 and 600 nm were obtained with excitation at 440, 410, 340, 420, 345 and 380 nm, respectively, in the presence of various amounts (0, 0.05, 0.1, 0.15, and 0.2 mg/ml) of the β-Lg and BSA (native, amyloid and amorphous aggregates)
The binding of the compounds to amorphous aggregate shows different behavior for different compounds, unlike the ThT and compound 1, the fluorescence intensity of the compounds 2, 3, 4 and 5 did not show the significant increasing upon interaction with the amorphous aggregate. From the curve slopes (Table 1), the increasing of fluorescence intensity was less significant in the presence of native protein/amorphous aggregate compare to amyloid aggregate (Fig. 4). Such steadily enhanced emission intensities are attributed to the less effective (or nonspecific) interaction between compounds (2–4) and native protein/amorphous aggregate. Based on the results obtained so far, we excluded compounds 1 and 5 from subsequent experiments, and measured the binding fluorescence of the compounds 2–4 and ThT to the amyloid/amorphous aggregate at different concentrations of the protein (0, 0.05, 0.1, 0.15, and 0.2 mg/ml) (Fig. 5). The binding fluorescence of compounds 2, 3 and 4 was demonstrated to be highly specific for amyloids as binding fluorescence was hardly observed with other proteins such as Globin (data not shown) [47,48,49] and amorphous protein aggregates made by bovine serum albumin (BSA) (Fig. 5). We then measured the curve slopes and the following orders were obtained for the dye-amyloid complexes and dye-amorphous complexes, respectively: 2 > 4 > ThT > 3, ThT > 2 > 4 > 3. This indicates that ThT binds to amorphous aggregate stronger than compounds 2–4, and compound 3 is more specific for distinguishing between the amyloid fibril and the amorphous shape. Therefore, it may be inferred that all tested compounds, especially 3 (with a specific configuration in aqueous solution) might experience a structural transition to new chromophores at the amyloid aggregate-bound state. Overall, these compounds appear to be reliable fluorescent probes to determine the amyloids quantitatively.

Comparison of binding fluorescence spectra between amyloid and amorphous aggregates. Synthetic compound 2–4 and ThT: compound 2 (filled triangle), compound 3 (open triangle), compound 4 (filled square), and ThT (open circle). The protein concentrations as previously mentioned, were considered: 0, 0.05, 0.1, 0.15, and 0.2 mg/ml for both amyloid and amorphous aggregates. Excitation and emission slit widths were set at 10–10. Further details are given in the “Material and methods” section
Fluorescence of ThT upon binding to amyloids has been suggested to vary depending on the dye concentration, and is quenched at high ThT concentrations [26]. In this study, β-Lg aggregation was assayed under the effect of various concentrations of compounds and the results were compared with the amyloid binding properties of ThT. The amyloid stock was prepared as mentioned earlier (“Material and methods”). The fluorescence of various amounts of ThT (at 483 nm) was evaluated in the presence of a fixed concentration (0.5 mg/ml) of amyloid aggregates. As shown in Fig. 6, the binding fluorescence was maximized at 7 µM of ThT and gradually decreased to 12 µM of the dye. This fact reflects that the fluorescence of free ThT molecules might be quenched by binding. Additionally, the dye concentration is responsible for the maximal binding fluorescence shifted to higher values as the amount of amyloids increased [for instance, see Fig. 6, compound 3, indicating that the optimal dye concentration actually depends on the quantity of amyloids analyzed]. In other words, an amyloid/dye ratio would be critical to obtaining an appropriate and optimal binding fluorescence signal. This would limit the universal use of ThT for amyloid detection. On the other hand, dye binding to the amyloid aggregates has been shown to exhibit an ideal property of saturation for a ligand interaction at the fixed concentration of the protein.

Scatchard plots of binding of the ThT and compound 3 to β-Lg amyloid aggregate. Changes in the dye binding fluorescence (a.u.) of β-Lg derived amyloid aggregates under the effect of the concentration of ThT and synthetic compound 3 were monitored. The β-Lg amyloid at 0.5 mg/ml were treated with various concentrations of either ThT or compound 3. The enhanced dye binding fluorescence intensities obtained at the corresponding maximum emission wavelengths and were plotted versus the dye concentrations. Data shown are representative example of three independent experiments and standard deviations were approximately within 5% of the experimental values. Further details are given in the paper text
Since the strong binding of the dye molecules is required for exact amyloid determination, there is a possibility that the fluorescence characteristic of each compound is controlled by the binding constants. To evaluate this possibility, we calculated dissociation constants using linear regression. It should be noted that, the compound 3 among other compounds was chosen, because it binds to amyloid aggregation more specific than other compounds and ThT, as we mentioned previously. For further investigation, Scatchard analyses for the binding of ThT and compound 3 to amyloid aggregation were measured and shown in Fig. 6. According to Table 2 and Fig. 6, which compare the Kd and Bmax of compound 3 with ThT, compound 3 had much higher binding affinity for amyloid aggregation than ThT. Furthermore, the initial slopes of the saturation curves (inside Fig. 6), which might reflect dependence of the compound binding fluorescence on the amount of dye, increased linearly as the amyloid level increased (Fig. 7). This indicates that the binding fluorescence is proportional not only to the compound concentration, but also the amount of amyloids. Therefore, it may be concluded that a relatively wide range of dye concentrations may be employed to detect amyloids, whereas the amount of ThT for an amyloid assay needs to be selected within a rather short range on the basis of the amyloid/dye ratio [26]. Taking the binding data into account, it can be concluded that compound 3 based on Table 2 and Fig. 6 may be considered as a potential fluorescent probe to quantify amyloid formation.

Two-dimensional representation of the Aβ peptide environment (interactions) in the binding site of the compounds 1–5 plus ThT (right); Molecular docking of the compounds 1–5 plus ThT (stick representation) in the main binding site (left). The ThT/compound binding take places at the interface between strands #1 and strands #2 [50]. Note: the same interacting side chains within the major ligand binding site revealed by molecular docking [51]
Although the fluorescence emission of the compounds 1 and 2 is less than ThT, and for 4 and 5, they are approximately equal to ThT, lack of the positive charge in these compounds is likely to help them pass through the membrane to detect amyloid aggregates within the cell as well as their antioxidant activity as opposed to ThT, make them good candidates both for detecting amyloid fibrils in vitro, in vivo and inhibiting the formation of the amyloid aggregates.
The antioxidant activity was assessed using two methods, including, 1,1-biphenyl-2-picrylhydrazyl (DPPH) radical scavenging [35], and ferric reducing antioxidant power (FRAP) assay [36, 37], according to the methods described in the “Material and methods”. The DPPH is a stable free radical with maximal absorption at about 520 nm, the DPPH radical has a deep violet colour in solution, and it becomes colourless or pale yellow (decrease in absorbance) when reduced by an antioxidant or a free radical species. This property allows visual monitoring of the reaction and determining the antioxidant activity of the tested substances with Trolox (2,5,7,8-tetramethychroman-2-carboxylic acid) as the standard [35]. The FRAP assay is a widely used method that provokes the antioxidant to behave as prooxidant in a redox-linked colorimetric reaction, wherein Fe3+ can be reduced by the antioxidant to an ion Fe2+. FRAP values are measured by comparing the change in absorbance at 593 nm in a test reaction mixture with that in mixtures containing ferrous ions at known concentrations. The DPPH and FRAP values of compounds are shown in Table 3. The antioxidant properties of all synthesized compounds except 5 were measured according to DPPH and FRAP methods [37]. Compounds 3 and 4 exhibited very good antioxidant properties that were more potent than Trolox as a reference compound. It seems that the antioxidant activity was increased when electron-donating groups such as hydroxyl and methoxy were added to the phenyl ring derived from aldehyde. This is due to the stabilization of the generated radical during oxidation. The compounds 3 and 4 have well antioxidative activity with a major activity for compound 4 (IC50 11.35 µM) in DPPH assay.
The results of antioxidant activity indicated that in addition to phenyl moiety, benzofuranone ring was also effective and important in this activity. However, it seems that during oxidation, benzofuranone moiety delocalized free electron. The result of DPPH assay was relatively consistent with that of reducing antioxidant power assay (FRAP). The potencies for the antioxidative activity of the synthesized compounds compared to the reference drug are in the following order: 4 > 3 > Trolox > 1 > 2. These results show that the synthesized compounds especially compound 3 could be a good candidate not only to detect the amyloid fibrils in in vitro, but also inhibit the toxicity of the aggregation by antioxidant activity.
A number of sensitive positron emission tomography (PET) tracers have been developed for amyloid imaging, but as indicated for ThT [18], most of them suffer from poor specificity and large signal to background ratio. So, design of tracers with specificity and improved binding affinity requires the knowledge about various potential binding sites in the Aβ/Tau fibrils available for the tracers and the nature of the local microenvironment of these sites. In this step of the work, we investigate potential binding sites of the compounds in the Aβ local structure of fibrils using the well-known probes, ThT. The molecular docking for β-Lg is not possible as the pdb file for aggregate form of β-Lg is not available on the pdb server. The binding affinity/profiles of the compounds (or probe candidates) and Thioflavin T to amyloid β fibrils (2beg) have been investigated by molecular docking, although the effect of fibril flexibility on the available binding sites was ignored. As indicated in Fig. 7, the probe ThT and the test compounds were found to bind at the same site of the fibril, within the fibril structure. The binding affinities for all compounds (and ThT as well) are found to be higher at the core sites than on the surface due to more contact residues (see Fig. 7). The binding affinity (docking score) of the tested molecules were in the following order: 3 > 4 > 2 > 1 > 5 > ThT, at a common site in the vicinity of Phe19, (site I [51], Fig. 7) which is in good agreement with experimental observations.
Since the synthesized phenolic compounds (PCs) and Thioflavin T can bind to amyloid fibrils in similar modes, some eligible PCs can be used to detect the presence of β-amyloid deposits, although due to the significant structural and binding affinity differences, they are used in quite different conditions. However, ThT can only be used as a fluorescent probe in vitro, since it cannot pass the BBB, PCs or those derivatives may serve as sensitive PET radiology and for the detection of amyloid fibrils both in vivo and in vitro.
Currently, there is no approved therapeutic agent directed towards the formation of amyloid aggregates. In the recent decades, application of several small phenolic compounds (PCs) [52] that efficiently inhibit the aggregation process and the associated cytotoxicity, has been considered as important therapeutic/preventive strategies [26, 53]. Regarding the radical scavenging activity of PCs, elevated oxidative stress as a major causative player in AD and the other neurodegenerative diseases [54], the inhibition mechanisms of PCs are mostly attributed to antioxidant activity, if these compounds cross the BBB and enter the central nervous system successfully.
On the other hand, protein aggregation is a hallmark of many neurodegenerative diseases, including Alzheimer’s and Parkinson’s. Although, the mechanisms linking aggregation to neurotoxicity remain poorly understood, partly because only limited information is available on the structure of these aggregates. Non-polar residues constitute approximately 30–50% of most proteins and play a critical role in aggregation characteristics of an amyloidogenic protein [55]. It is generally accepted that restricted geometry of planar aromatic stacking with proper directionality and orientation plays a critical role in acceleration of self-assembly processing of many cases of amyloid-related proteins (for instance, see [56]). Structural analyses [57] showed that Congo Red (CR), as an amyloid specific dye, interacts with stacked aromatic residues/β-strands within amyloidogenic interfaces, so it was also shown that CR can inhibit fibril formation as well.
Jung Sun Ahn et al. [58] reported that resveratrol, which may be classified either as a polyphenol or as a phytoestrogen, with the stilbene core structure, has potential to be used for quantitative determination of amyloid fibrils and as inhibitor as well. In continuation of our earlier work [26], we showed, in the current study, that the synthesized aromatic-rich compound 3, with fibril inhibition/antioxidant properties, are susceptible to be applied for detection/determination of amyloid assemblies.
Conclusion
In conclusion, we successfully designed and synthesized a new series of aurone derivatives as probes for imaging of amyloid plaques. In the in vitro binding studies, these aurones showed high binding affinity to Aβ aggregates based on selectivity and specificity parameters [13,14,15]. The development of molecules to be used as markers/probes of β-amyloid deposits in amyloid diseases has been a goal of researchers for several years [59]. Photophysical studies on the enhanced fluorescence behavior of ThT in the presence of extended β-structures confirm that the remarkable properties of the dye, as fluorescence marker of amyloid fibrils, are based on its behavior as molecular rotor [18]. Moreover, the existence of a wide solvent dependent distribution of ground-state conformations with very different photophysical behaviors makes ThT a highly complex (and non-specific) probe with strong wavelength dependencies of all its fluorescence properties. On the other hand, it has been frequently reported that ThT is unable to cross the BBB in vivo or to cross it in sufficient amounts for acceptable sensitivity [60, 61]; this is the result of its low lipophilicity and importantly, the presence of a permanent positive charge [62,63,64]. Uncharged aurone derivatives managed to permeate through the BBB in substantial amounts. Therefore, such a phenolic compounds without permanent positive charge and with better lipophilicity have been synthesized and they were probably able to cross BBB in vivo [60, 61, 65]. Two uncharged aurone derivatives, compounds 3 and 4 may be highly efficient in crossing the BBB, selective and specific binding to amyloid aggregates. According to our results, the synthetic compounds could be accounted as remarkable probes, but it should be investigated further as potential probes for detecting β amyloid aggregates in the AD brain.
Abbreviations
- AD:
-
Alzheimer’s disease
- MS:
-
Mass spectrometry
- ROS:
-
Reactive oxygen species
- ThT:
-
Thioflavin T
- TEM:
-
Transmission electron microscopy
- AFM:
-
Atomic force microscopy
- PMSF:
-
Phenyl methyl sulphonyl fluoride
- DMSO:
-
Dimethyl sulphoxide
- TFE:
-
2,2,2-Trifluoroethanol
- TMB:
-
3,3′,5,5′-Tetramethylbenzidine
- TMS:
-
Tetramethylsilane
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- β-Lg:
-
β-Lactoglobulin
- BSA:
-
Bovine serum albumin
- DPPH:
-
2,2-Diphenyl-1-picrylhydrazyl radical
- FRAP:
-
Ferric reducing antioxidant power
- PET:
-
Positron emission tomography
- PCs:
-
Phenolic compounds
- BBB:
-
Blood brain barrier
- CR:
-
Congo red
References
C.S. Sipe, M.R. Thomas, B.J. Stegmann, B.J. Van Voorhis, Hum. Reprod. 25, 690–696 (2010). https://doi.org/10.1093/humrep/dep442
T.P. Knowles, M. Vendruscolo, C.M. Dobson, Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014). https://doi.org/10.1038/nrm3810
F. Chiti, C.M. Dobson, Annu. Rev. Biochem. 75, 333–366 (2006). https://doi.org/10.1146/annurev.biochem.75.101304.123901
R.B. Maccioni, J.P. Munoz, L. Barbeito, Arch. Med Res. 32, 367–381 (2001)
J.A. Hardy, G.A. Higgins, Science 256, 184–185 (1992)
C. Vigo-Pelfrey, D. Lee, P. Keim, I. Lieberburg, D.B. Schenk, J. Neurochem. 61, 1965–1968 (1993)
D. Morgan, D.M. Diamond, P.E. Gottschall, K.E. Ugen, C. Dickey, J. Hardy, K. Duff, P. Jantzen, G. DiCarlo, D. Wilcock, K. Connor, J. Hatcher, C. Hope, M. Gordon, G.W. Arendash, Nature 408, 982–985 (2000). https://doi.org/10.1038/35050116
C.W. Cotman, E. Head, B.A. Muggenburg, S. Zicker, N.W. Milgram, Neurobiol. Aging 23, 809–818 (2002)
C.D. Smith, J.M. Carney, P.E. Starke-Reed, C.N. Oliver, E.R. Stadtman, R.A. Floyd, W.R. Markesbery, Proc. Natl. Acad. Sci. USA 88, 10540–10543 (1991)
W.R. Markesbery, Arch. Neurol. 56, 1449–1452 (1999)
D.A. Butterfield, J. Drake, C. Pocernich, A. Castegna, Trends Mol. Med. 7, 548–554 (2001)
C.M. Lauderback, J.M. Hackett, F.F. Huang, J.N. Keller, L.I. Szweda, W.R. Markesbery, D.A. Butterfield, J. Neurochem. 78, 413–416 (2001)
C. Behl, J.B. Davis, R. Lesley, D. Schubert, Cell 77, 817–827 (1994)
M.P. Mattson, Y. Goodman, Brain Res. 676, 219–224 (1995)
T. Pillot, B. Drouet, S. Queille, C. Labeur, J. Vandekerchkhove, M. Rosseneu, M. Pincon-Raymond, J. Chambaz, J. Neurochem. 73, 1626–1634 (1999)
K. Ono, T. Hamaguchi, H. Naiki, M. Yamada, Biochim. Biophys. Acta 1762, 575–586 (2006). https://doi.org/10.1016/j.bbadis.2006.03.002
O.S. Makin, L.C. Serpell, J. Mol. Biol. 335, 1279–1288 (2004)
S. Freire, M.H. de Araujo, W. Al-Soufi, M. Novo, Dyes Pigm. 110, 97–105 (2014). https://doi.org/10.1016/j.dyepig.2014.05.004
N. Darghal, A. Garnier-Suillerot, M. Salerno, Biochem. Biophys. Res. Commun. 343, 623–629 (2006)
R. Haudecoeur, A. Ahmed-Belkacem, W. Yi, A. Fortuné, R. Brillet, C. Belle, E. Nicolle, C. Pallier, J.-M. Pawlotsky, A. Boumendjel, J. Med. Chem. 54, 5395–5402 (2011)
M. Siah, M.H. Farzaei, M.R. Ashrafi-Kooshk, H. Adibi, S.S. Arab, M.R. Rashidi, R. Khodarahmi, Bioorg. Chem. 64, 74–84 (2016)
S.A. Ghadami, R. Khodarahmi, S. Ghobadi, M. Ghasemi, S. Pirmoradi, Biophys. Chem. 159, 311–320 (2011). https://doi.org/10.1016/j.bpc.2011.08.004
U.K. Laemmli, Nature 227, 680–685 (1970)
M.M. Bradford, Anal. Biochem. 72, 248–254 (1976)
M. Calamai, F. Chiti, C.M. Dobson, Biophys. J. 89, 4201–4210 (2005)
S.A. Ghadami, Z. Hossein-pour, R. Khodarahmi, S. Ghobadi, H. Adibi, Med. Chem. Res. 22, 115–126 (2013)
G. Giovannoni, J. Land, G. Keir, E. Thompson, S. Heales, Ann. Clin. Biochem. Int. J. Biochem. Med. 34, 193–198 (1997)
W. Wang, Int. J. Pharm. 289, 1–30 (2005)
K. Volkova, V. Kovalska, A. Balanda, R. Vermeij, V. Subramaniam, Y.L. Slominskii, S. Yarmoluk, J. Biochem. Biophys. Methods 70, 727–733 (2007)
M. Groenning, M. Norrman, J.M. Flink, M. van de Weert, J.T. Bukrinsky, G. Schluckebier, S. Frokjaer, J. Struct. Biol. 159, 483–497 (2007)
G.M. Morris, D.S. Goodsell, R. Huey, A.J. Olson, J Comput. Aided Mol. Des. 10, 293–304 (1996)
J.J. Esteb, L.M. McNulty, J. Magers, P. Morgan, A.M. Wilson, J. Chem. Educ. 87, 1074–1077 (2010)
M.D. Hanwell, D.E. Curtis, D.C. Lonie, T. Vandermeersch, E. Zurek, G.R. Hutchison, J. Cheminform. 4, 17 (2012). https://doi.org/10.1186/1758-2946-4-17
G.M. Morris, D.S. Goodsell, R.S. Halliday, R. Huey, W.E. Hart, R.K. Belew, A.J. Olson, J. Comput. Chem. 19, 1639–1662 (1998)
V. Bondet, W. Brand-Williams, C. Berset, LWT Food Sci. Technol. 30, 609–615 (1997). https://doi.org/10.1006/fstl.1997.0240
M.S. Blois, Nature 181, 1199–1200 (1958)
I.F. Benzie, J.J. Strain, Methods Enzymol. 299, 15–27 (1999)
M.G. Savelieff, A.S. DeToma, J.S. Derrick, M.H. Lim, Acc. Chem. Res. 47, 2475–2482 (2014). https://doi.org/10.1021/ar500152x
M. Ono, R. Watanabe, H. Kawashima, T. Kawai, H. Watanabe, M. Haratake, H. Saji, M. Nakayama, Bioorg. Med. Chem. 17, 2069–2076 (2009). https://doi.org/10.1016/j.bmc.2009.01.025
M. Ono, N. Yoshida, K. Ishibashi, M. Haratake, Y. Arano, H. Mori, M. Nakayama, J. Med. Chem. 48, 7253–7260 (2005). https://doi.org/10.1021/jm050635e
M. Hadjeri, C. Beney, A. Boumendjel, Curr. Org. Chem. 7, 679–689 (2003)
R. Khodarahmi, S.A. Karimi, M.R. Ashrafi Kooshk, S.A. Ghadami, S. Ghobadi, M. Amani, Spectrochim. Acta A Mol. Biomol. Spectrosc. 89, 177–186 (2012). https://doi.org/10.1016/j.saa.2011.12.058
P. Aymard, D. Durand, T. Nicolai, Int. J. Biol. Macromol. 19, 213–221 (1996)
J.M. Jung, G. Savin, M. Pouzot, C. Schmitt, R. Mezzenga, Biomacromolecules 9, 2477–2486 (2008). https://doi.org/10.1021/bm800502j
M.A.M. Hoffmann, P.J.J.M. van Mil, J. Agric. Food Chem. 45, 2942–2948 (1997). https://doi.org/10.1021/jf960789q
R. Eisert, L. Felau, L.R. Brown, Anal. Biochem. 353, 144–146 (2006). https://doi.org/10.1016/j.ab.2006.03.015
J.S. Kavanaugh, W.F. Moo-Penn, A. Arnone, Biochemistry 32, 2509–2513 (1993)
Y. Maya, M. Ono, H. Watanabe, M. Haratake, H. Saji, M. Nakayama, Bioconj. Chem. 20, 95–101 (2008)
M. Ono, Y. Maya, M. Haratake, K. Ito, H. Mori, M. Nakayama, Biochem. Biophys. Res. Commun. 361, 116–121 (2007)
R. Khodarahmi, M.R. Ashrafi-Kooshk, Int. J. Biol. Macromol. 100, 18–36 (2017). https://doi.org/10.1016/j.ijbiomac.2016.09.074
G. Kuang, N.A. Murugan, Y. Tu, A. Nordberg, H. Agren, J. Phys. Chem. B. 119, 11560–11567 (2015). https://doi.org/10.1021/acs.jpcb.5b05964
H. Rasouli, M.H. Farzaei, R. Khodarahmi, Int. J. Food Prop. 20, 1700–1741 (2017)
S.A. Ghadami, F. Bemporad, B.M. Sala, G. Tiana, S. Ricagno, F. Chiti, Cell. Mol. Life Sci. 74, 3577–3598 (2017)
W.J. Huang, X. Zhang, W.W. Chen, Biomed. Rep. 4, 519–522 (2016). https://doi.org/10.3892/br.2016.630
S. Maghsoudi, M.R. Ashrafi-Kooshk, M. Shahlaei, S.A. Ghadami, S. Ghobadi, A. Mostafaie, R. Khodarahmi, J. Iran. Chem. Soc. 10, 937–950 (2013). https://doi.org/10.1007/s13738-013-0231-7
A. Jangholi, M.R. Ashrafi-Kooshk, S.S. Arab, S. Karima, M. Poorebrahim, S.A. Ghadami, A.A. Moosavi-Movahedi, R. Khodarahmi, Int. J. Biol. Macromol. 109, 188–204 (2018). https://doi.org/10.1016/j.ijbiomac.2017.12.071
Y. Porat, A. Abramowitz, E. Gazit, Chem. Biol. Drug Des. 67, 27–37 (2006). https://doi.org/10.1111/j.1747-0285.2005.00318.x
J.S. Ahn, J.-H. Lee, J.-H. Kim, S.R. Paik, Anal. Biochem. 367, 259–265 (2007). https://doi.org/10.1016/j.ab.2007.05.023
W. Klunk, Neurobiol. Aging 19, 145–147 (1998)
Y. Li, X. Qiang, L. Luo, Y. Li, G. Xiao, Z. Tan, Y. Deng, Bioorg. Med. Chem. 24, 2342–2351 (2016)
K.-F. Liew, K.-L. Chan, C.-Y. Lee, Eur. J. Med. Chem. 94, 195–210 (2015)
W.E. Klunk, Y. Wang, G. Huang, M.L. Debnath, D.P. Holt, C.A. Mathis, Life Sci. 69, 1471–1484 (2001)
C.A. Mathis, B.J. Bacskai, S.T. Kajdasz, M.E. McLellan, M.P. Frosch, B.T. Hyman, D.P. Holt, Y. Wang, G.-F. Huang, M.L. Debnath, Bioorg. Med. Chem. Lett. 12, 295–298 (2002)
E.E. Nesterov, J. Skoch, B.T. Hyman, W.E. Klunk, B.J. Bacskai, T.M. Swager, Angew. Chem. Int. Ed. 44, 5452–5456 (2005)
J. Rautio, K. Laine, M. Gynther, J. Savolainen, AAPS J. 10, 92–102 (2008)
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Abbasbeigi, S., Adibi, H., Moradi, S. et al. Detection/quantification of amyloid aggregation in solution using the novel fluorescent benzofuranone-derivative compounds as amyloid fluorescent probes: synthesis and in vitro characterization. J IRAN CHEM SOC 16, 1225–1237 (2019). https://doi.org/10.1007/s13738-019-01599-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-019-01599-1