
Abstract
Neurodegenerative diseases are characterized by amyloid deposition. Thioflavin T (ThT) is one of the molecules considered for detection of amyloid deposits; however, its lipophilicity is too low to cross the blood–brain barrier. Therefore, there is a strong motivation to develop suitable compounds for in vitro fibril quantification as well as for in vivo amyloid imaging. Moreover, oxidative stress (particularly, uncontrolled peroxidase activity) has frequently been reported to play a critical role in the onset/progression of some neurodegenerative disorders. In this study, we describe the synthesis of some benzothiazole and benzofuranone compounds and examine their peroxidase inhibitory properties. Furthermore, to establish the potential binding of synthesized compounds to amyloid aggregates, their in vitro binding to some non-disease related amyloidogenic proteins were characterized. Analyses of the in vitro binding studies indicated that compounds 2 and 4 bind to the amyloid structures successfully while compounds 1 and 3 showed a low affinity in binding to fibrils. Furthermore, compounds 3 and 4 were observed to inhibit amyloid-mediated peroxidase activity in a reversible un-competitive manner.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amyloidosis defines a condition in which insoluble fibrillar protein aggregates are formed from innocuous soluble proteins; it is considered a common pathological feature of various neurodegenerative disorders, such as Alzheimer’s, Parkinson’s, Huntington’s, and Prion diseases (Bhak et al., 2009). For example, the characteristic signature of Alzheimer’s disease (AD) is the deposition of amyloid-β (Aβ) plaques and neurofibrillary tangles in the patient’s brain (Gorman and Chakrabartty, 2001), which parallel the disease but can only be diagnosed with certainty after death on autopsy. Amyloid structures and fibril formation (continuous β-sheet with the strands perpendicular to the longitudinal axis of fibril) (Jimenez et al., 1999; Sumner Makin and Serpell, 2004) have attracted a great deal of attention to reveal possible molecular reasons for the cytotoxicity, although their cause-or-effect relationship with the disorders is still elusive (Joseph et al., 2001; Robinson and Bishop, 2002). It has been suggested that protofibrillar intermediates are responsible for cell death by creating possible pore formation on cellular membranes, including the mitochondrial membrane (Mirzabekov et al., 1996; Lashuel et al., 2002; Murphy, 2007; Domanov and Kinnunen, 2008; Engel, 2009). Alternatively, amyloids may also provide a depot for redox-active metals that act as a production center for reactive oxygen species eventually affecting cell viability (Aliev et al., 2002; Takuma et al., 2005). Although known pathogenic proteins, such as Aβ are, typically involved in several neurodegenerative diseases, various non-disease-associated proteins can also be converted into amyloid fibrils (Darghal et al., 2006; Ghadami et al., 2011).
On the other hand, several researcher groups have recently proposed that Aβ and even other non-disease-related amyloid fibrils can potentially bind heme in vitro, and therefore, may induce oxidative stress in vivo. It is well accepted that oxidative stress to the central nervous system (CNS) manifests predominantly as lipid peroxidation as well as H2O2 formation. There are considerable amounts of intracellular peroxide species, free heme and amyloid fibrils present in the intracellular space; thus, the initiation of uncontrollable H2O2-dependent peroxidase activity of “heme-amyloid” system under the pathological conditions may damage intracellular DNA, neurotransmitters, and vital enzymes (as potential substrates) as a result of combined action of peroxides and peroxidase systems (Atamna and Frey, 2004; Atamna and Boyle, 2006; Atamna, 2006; Khodarahmi et al., 2009a, 2010). Also, since some anti-oxidant compounds display strong anti-fibrillogenic activity, a specific interconnection between oxidative stress (especially peroxide formation) and fibrillogenesis/neurodegeneration can be expected (Pratico et al., 1998; Ono and Yamada, 2006).
Amyloid formation has been monitored with various methods, including light scattering, Thioflavin T (ThT), Congo red, transmission electron microscopy (TEM), and atomic force microscopy (AFM) (Nilsson, 2004). Developing molecules to be used as markers of β-amyloid deposits in AD has been a goal of researchers for many years (Sumner Makin and Serpell, 2004). Although ThT binding fluorescence has been most widely employed to detect amyloids in vitro as well as ex vivo, the molecular mechanism of the dye binding to fibrils is not well established. Recently, it was suggested that enhanced ThT fluorescence originate from both dye-amyloid interaction and formation of ThT micelles on the fibrillar structures (Khurana et al., 2005). The binding fluorescence at 482 nm, on the other hand, would be quenched by unbound ThT present in excess during the amyloid assay.
The need for new analytical tools is highlighted with regard to the fact that a comprehensive understanding of molecular details of amyloidosis is essential for developing strategies to control amyloid formation and consequently, neurodegenerative disorders, and this requires a more reliable probe for identifying amyloids. Since compounds without a permanent positive charge are mainly capable of crossing the blood–brain barrier (BBB) (Darghal et al., 2006), in this study, we synthesized and employed benzothiazole and benzofuranone derivatives (including neutral ThT analogues), both as fluorescent probes to quantitatively determine the amyloid fibrils made of chymotrypsin (Khodarahmi et al., 2009b) and crystallin (Khodarahmi et al., 2010) and as potential inhibitors for peroxidase activity. The resulting data may be useful in providing mechanistic insights to develop potential diagnostic, curative, and/or preventive strategies in vivo against amyloid-related neurodegenerative disorders.
Materials and methods
Materials and equipments
3,3′,5,5′-tetramethylbenzidine (TMB), 30 % H2O2, PMSF, bovine α-chymotrypsin, and ThT were obtained from Sigma chemical company (St. Louis, MO, USA). TFE was purchased from Merck (Darmstadt, Germany). All other chemicals were of the highest analytical grade of purity available and were used as obtained from suppliers. All solutions were prepared with double distilled water. Unless otherwise stated, all solutions were made in 20 mM sodium phosphate buffer (pH 7.4). A stock solution of ThT (1 mM) was freshly prepared in water and stored in dark at 4 °C. A Cary-100 Bio (VARIAN) spectrophotometer was used for protein determination and peroxidase assay. All fluorescence measurements were performed in the ratio mode using a 1 cm cell in a Cary Eclipse (VARIAN) fluorescence spectrophotometer, equipped with a 150 W xenon lamp and a thermostated cell holder, at room temperature or as stated. Appropriate vehicle controls were run in all experiments. All the reported results are averages of 2–3 separate experiments whenever the coefficients of variation were less than 5 %.
Chemistry
All chemicals were purchased from Merck and Fluka chemical companies. The products were characterized by comparing the physical data with those of known samples or by their spectral data. Column chromatography was performed using silica gel 60 (230–400 mesh). All yields refer to isolated yield. The chemical structures of test compounds are summarized in Fig. 1. The structure of compounds was characterized by IR, 1H NMR, and 13C NMR spectra. The melting point was taken on a Kofler hot stage apparatus and is uncorrected. IR spectrum was recorded on a Shimadzu 470 spectrophotometer (KBr disk). 1H NMR spectrum was recorded on a Bruker FT-200 NMR spectrophotometer using CDCl3 as solvent and TMS as an internal standard. The purity of the compound was monitored by thin layer chromatography.

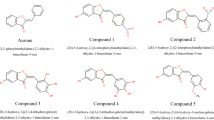
Chemical structures of synthesized compounds (2-(4-methoxyphenyl)benzo[d]thiazole, 1, 4-(benzo[d]thiazol-2-yl)-N,N-dimethylaniline, 2, (E)-6-hydroxy-2-(2-hydroxy-3-methoxybenzylidene)benzofuran-3(2H)-one, 3 and (E)-2-(2,4-dihydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one, 4. See also ThT and resveratrol structures (center) for comparison
Typical procedure for synthesis of (2-(4-methoxyphenyl)benzo[d]thiazole) (compound 1) and (4-(benzo[d]thiazol-2-yl)-N,N-dimethylaniline) (compound 2) as known compounds (see “Bahrami et al., 2008” and also Fig. 1).
In a round-bottomed flask (50 mL) equipped with a magnetic stirrer, a solution of o-aminothiophenol (1 mmol), 4-methoxybenzaldehyde, and/or 4-N,N-dimethylaminobenzaldehyde (1 mmol) in MeCN (3 mL) was prepared. H2O2 (30 %, 4 mmol, 0.4 mL) and NH4Ce(NO2)6 (0.1 mmol, 0.0548 g) were added and the mixture was stirred at room temperature for 1 h. The progress of the reaction was monitored by TLC (eluent: n-hexane/ethyl acetate: 6/4). When the starting materials had completely disappeared, the reaction mixture was quenched by adding water (10 mL), extracted with ethyl acetate (4 × 10 mL) and the extract dried with anhydrous MgSO4. The filtrate was evaporated and the corresponding benzothiazole was obtained as the only product (Bahrami et al., 2008).
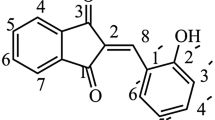
(E)-6-hydroxy-2-(2-hydroxy-3-methoxybenzylidene)benzofuran-3(2H)-one (compound 3) and (E)-2-(2,4-dihydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one, (compound 4).
In order to synthesize (E)-6-hydroxy-2-(2-hydroxy-3-methoxybenzylidene)benzofuran-3(2H)-one (compound 3) and (E)-2-(2,4-dihydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one, (compound 4, see Fig. 1), as new compounds, the starting 6-hydroxy-3-coumaranone B (see Scheme 1) was prepared by dissolving resorcinol (1 mmol) and bromoacetyl bromide (1 mmol) in acetonitrile (3 mL) with 400 mg silica sulfuric acid added as catalyst. The reaction mixture was heated at 80 °C for 3 h. After cooling to room temperature and filtering the reaction mixture, the compound A was purified by column chromatography. Subsequently, the compound A was cyclized using NaOH 2 M to produce 6-hydroxy-3-coumaranone B in 75 % yield after 1 h. mp = 243–246 °C; 1H NMR (CDCl3) δ 7.411 (d, 1H, H5), 6.542–6.568 (d, 1H, H4), 6.664 (s, 1H, H7), 4.602 (s, 2H, CH2), 4.580 (s, 1H, OH); IR (KBr, cm−1) υ 1745 (C=O), 3452 (OH).

Synthesis of compounds 3 and 4: reagents and conditions
To a solution of 6-hydroxy-3-coumaranone B (1 mmol), and appropriate aryl aldehyde (1 mmol) in ethanol (3 mL) was added piperazine (0.1 mmol) as the basic catalyst. The reaction mixture was heated in 80 °C for 2 h. The reaction mixture was allowed to stand at room temperature for 24 h. The precipitate was filtered, dried, and crystallized from acetic acid to afford pure (E)-6-hydroxy-2-(2-hydroxy-3-methoxybenzylidene) benzofuran-3(2H)-one 3 and (E)-2-(2,4-dihydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one 4 in 70 and 65 % yields, respectively (Scheme 1). Structural assignments of the products are based on their IR, 1H NMR, 13C NMR spectra, and melting point.
Compound 3: (E)-6-hydroxy-2-(2-hydroxy-3-methoxybenzylidene) benzofuran-3(2H)-one 3: yield 70 %; mp = 305–306 °C; 1H NMR (CDCl3) δ 7.71 (d, 1H, H 4), 7.62 (s, 1H, C=CH), 7.11–7.17 (m, 2H, H 5′, H 6′), 7.04 (s, 1H, H7), 6.91 (s, 1H, H 7), 6.73 (d, 1H, H ‚4 ), 6.50 (d, 1H, H 5), 5.22 (s, 2H, OH), 3.84 (s, 3H, OCH 3); 13C NMR (CDCl3) δ 182.1, 167.5, 165.2, 151.2, 148.4, 146.7, 128.0, 122.6, 121.4, 117.4, 115.2, 111.8, 105.3, 101.3, 56.4; IR (KBr, cm−1) υ 1730 (C=O), 3250 (OH).
Compound 4: (E)-2-(2,4-dihydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one 4: yield 65 %; mp = 330–332 °C; 1H NMR (CDCl3) δ 7.96–7.90 (m, 2H, H 6′, H 4), 7.35 (s, 1H, C=CH), 6.70 (s, 1H, H 7), 6.55 (d, 1H, H 5), 6.25 (d, 1H, H 5′), 6.15 (s, 1H, H3′), 3.49–3.42 (s, 3H, OH); 13C NMR (CDCl3) δ 182.1, 167.0, 165.2, 159.3, 157.3, 146.7, 131.3, 128.3, 115.2, 112.3, 108.3, 105.7, 103.5, IR (KBr, cm−1) υ 1735 (C=O), 3455 (OH).
Sample preparation and fibrillogenesis
Amyloid made of α-chymotrypsin was monitored with the dyes binding fluorescence. The α-chymotrypsin, was converted to amyloid-like fibrils in the presence of 32 % TFE (Khodarahmi et al., 2009b, Nilsson, 2004).
Preparation of stock solutions of dyes and biological molecules
1 × 10−3 M dye stock solutions were prepared by dissolving the dye in DMSO. The concentrations of proteins in stock solutions were 2 mg/mL for both native and fibrillar proteins. The fibrils used in all experiments came from the same batch.
Preparation of working solutions
Working solutions of free dyes at 5 × 10−6 M were prepared by dilution of the dye stock solution in 20 mM sodium phosphate buffer (pH 7.4). Working solutions of dye–protein complexes were prepared by mixing an aliquot of the dye stock solution (2.0 μL) with an aliquot (1, 2, and 3 μL) of native or fibrillar proteins in buffer. Concentrations of the proteins in working solution were 0.03, 0.05, 0.07, 0.10, and 0.30 mg/ml, respectively for both native and aggregated proteins. Measurements were performed, in the presence of 10 μL of protein, only for the most efficient dyes which demonstrated significant fibrillar proteins binding preference values as compared with native protein. All working solutions were prepared immediately before the experiment.
Spectroscopic measurements
Fluorescence excitation and emission spectra were collected on a Cary Eclipse fluorescence spectrophotometer (Varian, Australia). Fluorescence spectra were measured with excitation and emission slit widths set to 10 nm. The correction of the obtained spectra for the wavelength dependence on the excitation source intensity was performed by the Cary Eclipse fluorescence spectrophotometer. At the same time, fluorescence excitation and emission spectra were not corrected for the sensitivity of the Cary Eclipse detection system. However, the primary aim of the work was to study the fluorescent response of each dye in the presence of native and aggregated proteins. Since for each dye the positions of the corresponding spectra are very similar in free form and in the presence of native and aggregated proteins, the absence of correction for the detection system sensitivity does not influence the results of our study. In vitro binding studies were conducted at least in triplicate by the procedures described in detail by Groenning et al. (2007). Spectroscopic measurements were performed in standard quartz cells (1 × 1 cm). All measurements were carried out at room temperature. All measurements were made at the respective excitation maxima of each dye.
Peroxidase activity of the horseradish peroxidase
The peroxidase activity of horseradish peroxidase (HRP) was measured by oxidation of TMB by H2O2 by following the increase in absorbance at 652 nm, which allows continuous monitoring of the TMB oxidation product. The peroxidase activity of horseradish peroxidase (0.07 μg/mL) was tested in the concentration range of 0.3–5.0 mM of the TMB for 60 s at room temperature. The effect of potential inhibitor compounds on peroxidase activity was measured by TMB assay. The extent of TMB oxidation by horseradish peroxidase in the absence or presences of compounds were tested in the kinetic mode for 60 s at room temperature and the values of ΔA/Δt were determined.
Results and discussion
Amyloid formation has been monitored with various spectroscopic methods, using ThT and congo red (CR). In addition, the resveratrol (the major phenolic constituent of red wine) binding fluorescence was recently demonstrated to be highly specific for the amyloids (Ahn et al., 2007). Thus, there is a possibility that other phenolic and or thiazole compounds show selectivity in binding to amyloid fibrils and are possibly applicable in both in vivo amyloid imaging techniques and in situ staining of amyloid plaques in tissue section.
Binding of compounds 1–4 to the α-chymotrypsin amyloid aggregate was examined by observing fluorescence emission spectra between 300 and 600 nm using specific excitation wavelengths (360, 400, 398, and 408 nm for compounds 1–4, respectively) and at a fixed concentration (40 μM) of compounds. Also, the excitation wavelength for ThT was set at 442 nm. As indicated in Fig. 2, the fluorescence intensities (emissions at 400, 430, 500, and 500 nm, respectively) increased significantly upon interaction with amyloid aggregates. Moreover, as the amount of amyloid aggregates (corresponding to the concentration of native α-chymotrypsin solution) increased, the fluorescence intensities were augmented proportionally so that the maximum fluorescence emissions increased linearly as a function of amyloid concentration. Unlike the amyloid aggregates, however, the fluorescence intensity of the test compounds did not undergo a significant change upon interaction with the soluble (monomeric) form of α-chymotrypsin. As is evident from the curve slopes (Fig. 3), the increase in dye fluorescence was less significant in the presence of native protein. Such mild and steadily enhanced emission intensities are attributed to the less effective (or nonspecific) interaction between compounds and amyloid fibrils. Therefore, it may be inferred that all tested compounds, especially X and Y (with a specific configuration in aqueous solution) might experience structural transition to new chromophores at the amyloid aggregate-bound state. Overall, these compounds appear to be reliable fluorescent probes to determine the amyloids quantitatively. In addition, the binding fluorescence of test compounds was demonstrated to be highly specific for amyloids as binding fluorescence was hardly observed with other proteins such as globin (a protein with 8 consecutive α-helical segments, (Kavanaugh et al., 1993, see also Fig. 3) and native crystalline solution (and amorphous protein aggregates made of chymotrypsin, data not shown). As demonstrated by the curve slopes (Fig. 3), the increase in fluorescence intensities was much less significant compared to those observed for dye–amyloid interaction. These steadily decreasing emission intensities may be attributed to generation of microenvironments with low dielectric constant (within or between soluble protein molecules and) upon amorphous protein aggregation. Selective binding to amyloid-containing aggregates may make the test compound possibly applicable in imaging and in situ staining of amyloid fibrils.

Binding fluorescence spectra of test compounds in the presence of α-chymotrypsin amyloid aggregates. Compounds 1–4 binding fluorescence spectra between 300 and 600 nm were obtained with excitation at 360, 400, 398, and 408 nm, respectively, in the presence of various amounts (0, 0.03, 0.05, 0.07, 0.1, and 0.32 mg/ml) of the α-chymotrypsin aggregates (open circle) and were plotted separately with various amounts of the native protein (open square). The fluorescence intensities were plotted with a fixed amount of compounds at 40 μM. Data shown are representative example of three independent experiments and standard deviations were approximately within 5 % of the experimental values. Further details are given in the “Material and methods” section

Fluorescence characteristics of test compounds under the effect of various amounts of α-chymotrypsin amyloid aggregates (filled triangle), native α-crystallin (open square), and heme-free globin (open triangle). The protein concentrations were 0, 0.03, 0.05, 0.07, 0.10, and 0.32 mg/mL in 50 mM phosphate buffer (pH 7.4); in the presence of fixed amount (10 mM) of compounds. The binding fluorescences were monitored with the emission scan between 300 and 600 nm following excitations at 360, 400, 398, and 408 nm, respectively. Ribbon representation of human oxyhemoglobin (left) and bovine AlphaA crystallin (right) were obtained from the Protein Data Bank (ID code 1HHO and 3L1E, respectively). The structures visualization and figures generation were performed with PyMOL version 0.99 beta06 (http://www.pymol.org) (Fendri et al., 2007). Data shown are representative example of three independent experiments and standard deviations were approximately within 5 % of the experimental values. Further details are given in the “Material and methods” section
Scaria et al. (1986) studied the effect of dye binding on the conformation of poly l-lysine polypeptides. They observed that dye binding leads to a conformational transition of poly l-lysine from the α-helix to the β-sheet, the structure with further susceptibility for binding to dye molecules. Therefore, it can be concluded that strong interaction between dyes molecules and β-sheet structures is more likely to be due to geometric considerations. In addition, considering the presence of extended β-conformers in amyloid structures (see scheme 2), further binding of dye molecules may be anticipated. Thus, we observed a large difference in dye fluorescence characteristics between “dye-native crystalline” (a β-rich protein, see Fig. 3) and “dye-amyloid aggregate” incubation mixtures (Fig. 3).

The common structure of amyloid fibrils and a structural rationale for fibril–dye interactions. Top cross-β structure of amyloid fibrils, formed from layers of laminated β-sheets. Bottom “Channel” model of dye binding to fibril-like β-sheets. Dye is proposed to bind along surface side-chain grooves running parallel to the long axis of the β-sheet (See Khodarahmi et al., 2010 and reference [38] within. Also, for better interpretation, the reader is referred to the web version of this article)
Although the exact mechanism of dye binding to amyloid fibrils remains unclear and, putting aside the report of (Eisert et al., 2006), most of the binding models described in the literature state that binding of amyloid-specific dyes (such as ThT and Congo red) occurs along their long molecular axis parallel to the fibril axis (Krebs et al., 2005; Cooper, 1974; Jin et al., 2003). As the side chains on each side of beta-sheet form neat rows (so called “channels”, see also scheme 2) running along the fibril (and therefore perpendicular to the strands), it seems likely that the dye molecule can insert itself into these channels. Thus, the dye molecule must be flat and thin enough to enter such a binding channel (Krebs et al., 2005). Based on this data, it may be expected that bulky substituents in the dye structures will complicate dye insertion in this channel. On the other hand, different fluorescent dyes used for amyloid detection are proposed to bind to fibrils via diverse binding sites (Volkova et al., 2007). Due to the mentioned reasons, binding mechanism of dyes specific for amyloid fibrils requires further investigation.
ThT binding fluorescence of amyloids has been suggested to vary depending on the dye concentration so that its binding fluorescence is quenched at high dye concentrations. In this study, α-chymotrypsin amyloidogenesis was assayed under the effect of various concentrations of compounds and the results were compared with the amyloid binding properties of ThT. The amyloid stock was prepared by incubating 2 mg/mL of α-chymotrypsin, in the presence of 32 % TFE. The fluorescence of various amounts of ThT (at 482 nm) was evaluated in the presence of a fixed concentration (0.1 mg/mL) of amyloid aggregates. As indicated by Fig. 4, the binding fluorescence was maximal at 80 μM of ThT and gradually decreased to 200 μM of the dye. This fact reflects that the free ThT molecules might quench the binding fluorescence.

Changes in the dye binding fluorescence (a.u.) of α-chymotrypsin-derived amyloid aggregates under the effect of the concentration of (ThT and) compounds. The α-chymotrypsin amyloids at three different concentrations of 0.02 (open square), 0.05 (open circle), and 0.1 (open triangle) mg/mL were treated with various concentrations of either ThT or compounds. The enhanced dye binding fluorescence intensities obtained at the corresponding maximum emission wavelengths and were plotted versus the dye concentrations
Also, the dye concentration responsible for the maximal binding fluorescence shifted to higher values as the amount of amyloids increased (for instance, see Fig. 4, compound 3), indicating that the optimal dye concentration actually depends on the quantity of amyloids analyzed. In other words, an amyloid/dye ratio would be critical to obtaining an appropriate and optimal binding fluorescence signal. This would limit universal use of ThT for amyloid detection. On the other hand, dye binding to the amyloid aggregates has been shown to exhibit an ideal property of saturation for a ligand interaction at the fixed concentration of the protein (Fig. 4). Since strong binding of the dye molecules is required for exact amyloid determination, there is a possibility that fluorescence characteristic of each compound is controlled by the binding constants. To evaluate this possibility, we calculated dissociation constants using linear regression. According to Fig. 5, which compares the K d and B max for compound as well as ThT, Scatchard analyses for the binding of compounds 1–4 showed apparent K d = 35.2, 2.6, 16.9, 3.0 (μM) and B max = 59.9, 30.6, 93.2, 12.1 (μM dye/μM amyloid), respectively. The calculated dissociation constants for dye–amyloid complexes are in the following order: 1 > 3 > 4 > 2. Furthermore, the initial slopes of the saturation curves, which might reflect dependence of the compounds binding fluorescence on the amount of compounds, increased linearly as the amyloid level rose (Fig. 5). This indicates that the binding fluorescence is proportional to not only the compound concentration but also the amount of amyloids. Therefore, it may be concluded that a relatively wide range of dye concentrations may be employed to detect amyloids, whereas the amount of ThT for an amyloid assay needs to be selected within a rather short range on the basis of the amyloid/dye ratio. Taking the binding data into account, it can be concluded that compounds 2, 4 may be considered as potential fluorescent probes to quantify amyloid formation.

Scatchard plots of binding of the synthesized compounds to α-chymotrypsin amyloid aggregates. Data shown are representative example of three independent experiments and standard deviations were approximately within 5 % of the experimental values. Further details are given in the paper text
The development of molecules to be used as markers/probes of β-amyloid deposits in amyloid diseases has been a goal of researchers for several years (Klunk, 1998). ThT is one of the (candidate) molecules of choice in attempts made at detection of amyloid deposits in ex vivo and in vitro systems. This compound is a histologic benzothiazole dye (see Fig. 1) capable of staining amyloid structures selectively. It has been frequently reported that ThT is unable to cross BBB in vivo or to cross it in sufficient amounts for acceptable sensitivity; this is the result of its low lipophilicity and importantly, the presence of a permanent positive charge (Klunk et al., 2001; Mathis et al., 2002; Nesterov et al., 2005). Uncharged analogues of ThT managed to permeate through the BBB in substantial amounts (see compounds 1 and 2 structures in current study). Therefore, ThT derivatives without permanent positive charge and with better lipophilicity (but not excessively, as this will lead to its aggregation and accumulation on blood proteins and within red blood cell membranes) have been synthesized and they were reportedly able to cross BBB in vivo (Mathis et al., 2002; Nesterov et al., 2005). For example, two ThT uncharged analogues, compounds 1 and 2 may be highly efficient both in crossing the BBB and in selective binding to amyloid aggregates. For general use, the binding of these potential compounds to amyloids needs to be further evaluated in terms of its β-sheet interaction and a possible formation of new chromophore on its amyloid binding. In addition, other practical applications of the candidate compounds, such as the ability to detect amyloids within tissues, are worth investigating.
Despite extensive efforts around the world, development of an effective treatment for neurodegenerative diseases such as AD remains elusive. Current therapeutic strategies are limited to those that attenuate AD symptomology without deterring the progress of the disease itself, and thus only postpone the inevitable deterioration of the affected individual. Notably, as oxidative stress is perhaps the earliest feature of an AD brain (Nunomura et al., 2001; Zhu et al., 2001, 2004), successful neuronal protection from oxidative damage will potentially prevent the disease altogether, if appropriately administered. In this regard, efforts focused on elucidation of the molecular mechanisms involved in the oxidative stress within the cell have yielded significant results and will hopefully succeed through clinical trials for efficacy. Recently, it has been proposed that Aβ, a pathogenic peptide in AD, can potentially bind heme, forming an Aβ-heme complex, in vitro (Howlett et al., 1997; Atamna and Boyle, 2006). In this case, the binding of heme to protein creates heme deficiency, mitochondrial dysfunction and increased production of oxidants such as H2O2 (Atamna and Boyle, 2006; Atamna and Frey, 2004). Increasing evidence suggests that hydrogen peroxide and possibly other hydroperoxide species are generated during the very early stages of amyloid formation and that oxidative stress plays an important role in the pathogenesis of this type of neurodegenerative disorder (Khodarahmi et al., 2009a; Turnbull et al., 2003). Oxidative stress to the CNS (and to the other organs) manifests predominantly as lipid peroxidation due to the susceptibility of unsaturated fatty acids to oxidation (Butterfield et al., 2002). Despite extensive experimental work conducted in this field, the mechanism involved in the peroxide release is still not completely understood. Furthermore, there is discrepancy between different reports on chronological precedence of amyloid aggregation and oxidative reactions. On the other hand, Atamna (2006) and Khodarahmi et al. (2010) reported the H2O2-dependent peroxidase activity of “heme-amyloidogenic protein” complex using in vitro experimental systems. Due to the presence of high amounts of peroxide species other than H2O2, in some amyloid diseases (Pratico et al., 1998), there is a possibility that the generated peroxide molecules serve as oxidant substrate for peroxidase systems. Also, since anti-oxidant compounds have potent anti-fibrillogenic and fibril destabilizing effects in a dose–dependent manner (Ono and Yamada, 2006), a specific interconnection between peroxide formation and fibrillogenesis can be expected. Since the basic and clinical aspects of heme binding to (structural intermediates of) amyloid fibrils as well as observed peroxidase activity (Pramanik and Dey, 2011; Yuan et al., 2012) have yet to be identified, additional data on this field may provide insight to manage amyloid-related processes/diseases. Considering the potential role of peroxidase activity of “heme-amyloid fibril” complex in onset/progression of neurodegenerative disorders, as well as the fact that peroxidase-mediated oxidative stress can play at least a partially causative rather than merely by-stander role in the neurofibrillary pathology in AD/PD in vivo, and may affect nucleic acids, enzymes, and neurotransmitters (Yuan et al., 2012), ongoing clinical trials should therefore focus on simultaneous application of fibril stabilizers, aggregation inhibitors and peroxidase inhibitors as a new generation of amyloid-based therapeutics. In the current study, since phenolic compounds 3 and 4 showed significant anti-oxidant activity using ferric reducing antioxidant power (FRAP), oxygen radical absorbance capacity (ORAC), and 1,1-biphenyl-2-picrylhydrazyl (DPPH) methods (data not shown), they were considered as potential inhibitors of peroxidase activity. The peroxidase activity of HRP, at different TMB concentrations, either in the presence or absence of compounds 3 and 4 were assessed and the results were appraised using Lineweaver–Burk plots. TMB, a classic substrate for peroxidases, was rapidly oxidized by H2O2 when enzyme was added. Clearly, peroxidase activity follows a linear kinetic pattern with no significant lag period as demonstrated by the increase in absorbance, at 652 nm (data not shown). Then, enzyme activity was followed in the presence of 0, 50, and 150 mM of compounds 3, 4 (and 2.6 mM H2O2). As indicated in Fig. 6, it was found that both compounds inhibit the HRP peroxidase activity through an un-competitive mechanism. It is noteworthy that due to the heterogeneous nature and inherent turbidity of insoluble amyloid aggregates, changes in the peroxidase activity in the presence of the compounds, was recorded using native peroxidase enzyme (HRP). Previous studies (Atamna and Frey, 2004; Atamna and Boyle, 2006; Khodarahmi et al., 2010) have indicated the similar catalytic behavior of “amyloid-heme” complex and the native peroxidase.

The un-competitive inhibition of HRP activity. Lineweaver–Burk plot for the inhibition of peroxidase activity by compounds 3 (left) and 4 (right). The enzyme concentration in the assay buffer (potassium phosphate 20 mM, pH 7.2) was 0.7 × 10−4 mg/mL. The units of Y and X axes are (min.Abs−1) and (mM−1), respectively
Abbreviations
- PMSF:
-
Phenylmethylsulphonyl fluoride
- DMSO:
-
Dimethyl sulphoxide
- TFE:
-
2,2,2-Trifluoroethanol
- ThT:
-
Thioflavin T
- TMB:
-
3,3′,5,5′-Tetramethylbenzidine
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- Aβ:
-
Amyloid-β
- CNS:
-
Central nervous system
- BBB:
-
Blood–brain barrier
- TEM:
-
Transmission electron microscopy
- AFM:
-
Atomic force microscopy
- HRP:
-
Horseradish peroxidase
- DPPH:
-
1,1-Biphenyl-2-picrylhydrazyl
- FRAP:
-
Ferric reducing antioxidant power
- ORAC:
-
Oxygen radical absorbance capacity
References
Ahn JS, Lee JH, Kim JH, Paik SR (2007) Novel method for quantitative determination of amyloid fibrils of alpha-synuclein and amyloid beta/A4 protein by using resveratrol. Anal Biochem 367:259–265
Aliev G, Smith MA, Seyidov D, Neal ML, Lamb BT, Nunomura A, Gasimov EK, Vinters HV, Perry G, LaManna JC, Friedland RP (2002) The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s disease. Brain Pathol 12:21–35
Atamna H (2006) Heme binding to amyloid-beta peptide: mechanistic role in Alzheimer’s disease. J Alzheimers Dis 10:255–266
Atamna H, Boyle K (2006) Amyloid-beta peptide binds with heme to form a peroxidase: relationship to the cytopathologies of Alzheimer’s disease. Proc Natl Acad Sci USA 103:3381–3386
Atamna H, Frey WH 2nd (2004) A role for heme in Alzheimer’s disease: heme binds amyloid beta and has altered metabolism. Proc Natl Acad Sci USA 101:11153–11158
Bahrami K, Khodaei MM, Naali F (2008) Mild and highly efficient method for the synthesis of 2-arylbenzimidazoles and 2-arylbenzothiazoles. J Org Chem 73:6835–6837
Bhak G, Choe YJ, Paik SR (2009) Mechanism of amyloidogenesis: nucleation-dependent fibrillation versus double-concerted fibrillation. BMB Rep 42:541–551
Butterfield DA, Castegna A, Lauderback CM, Drake J (2002) Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging 23:655–664
Cooper JH (1974) Selective amyloid staining as a function of amyloid composition and structure. Histochemical analysis of the alkaline Congo red, standardized toluidine blue, and iodine methods. Lab Invest 31:232–238
Darghal N, Garnier-Suillerot A, Salerno M (2006) Mechanism of thioflavin T accumulation inside cells overexpressing P-glycoprotein or multidrug resistance-associated protein: role of lipophilicity and positive charge. Biochem Biophys Res Commun 343:623–629
Domanov YA, Kinnunen PK (2008) Islet amyloid polypeptide forms rigid lipid-protein amyloid fibrils on supported phospholipid bilayers. J Mol Biol 376:42–54
Eisert R, Felau L, Brown LR (2006) Methods for enhancing the accuracy and reproducibility of Congo red and thioflavin T assays. Anal Biochem 353:144–146
Engel MF (2009) Membrane permeabilization by islet amyloid polypeptide. Chem Phys Lipids 160:1–10
Fendri A, Frikha F, Miled N, Ben Bacha A, Gargouri Y (2007) Modulating the activity of avian pancreatic lipases by an alkyl chain reacting with an accessible sulfhydryl group. Biochem Biophys Res Commun 360:765–771
Ghadami SA, Khodarahmi R, Ghobadi S, Ghasemi M, Pirmoradi S (2011) Amyloid fibril formation by native and modified bovine beta-lactoglobulins proceeds through unfolded form of proteins: a comparative study. Biophys Chem 159:311–320
Gorman PM, Chakrabartty A (2001) Alzheimer beta-amyloid peptides: structures of amyloid fibrils and alternate aggregation products. Biopolymers 60:381–394
Groenning M, Norrman M, Flink JM, van de Weert M, Bukrinsky JT, Schluckebier G, Frokjaer S (2007) Binding mode of thioflavin T in insulin amyloid fibrils. J Struct Biol 159:483–497
Howlett D, Cutler P, Heales S, Camilleri P (1997) Hemin and related porphyrins inhibit beta-amyloid aggregation. FEBS Lett 417:249–251
Jimenez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M, Saibil HR (1999) Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO J 18:815–821
Jin LW, Claborn KA, Kurimoto M, Geday MA, Maezawa I, Sohraby F, Estrada M, Kaminksy W, Kahr B (2003) Imaging linear birefringence and dichroism in cerebral amyloid pathologies. Proc Natl Acad Sci USA 100:15294–15298
Joseph J, Shukitt-Hale B, Denisova NA, Martin A, Perry G, Smith MA (2001) Copernicus revisited: amyloid beta in Alzheimer’s disease. Neurobiol Aging 22:131–146
Kavanaugh JS, Moo-Penn WF, Arnone A (1993) Accommodation of insertions in helices: the mutation in hemoglobin Catonsville (Pro 37 alpha-Glu-Thr 38 alpha) generates a 3(10)--> alpha bulge. Biochemistry 32:2509–2513
Khodarahmi R, Soori H, Karimi SA (2009a) Chaperone-like activity of heme group against amyloid-like fibril formation by hen egg ovalbumin: possible mechanism of action. Int J Biol Macromol 44:98–106
Khodarahmi R, Soori H, Amani M (2009b) Study of cosolvent-induced alpha-chymotrypsin fibrillogenesis: does protein surface hydrophobicity trigger early stages of aggregation reaction? Protein J 28:349–361
Khodarahmi R, Naderi F, Mostafaie A, Mansouri K (2010) Heme, as a chaperone, binds to amyloid fibrils and forms peroxidase in vitro: possible evidence on critical role of non-specific peroxidase activity in neurodegenerative disease onset/progression using the alpha-crystallin-based experimental system. Arch Biochem Biophys 494:205–215
Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, Roy R, Singh S (2005) Mechanism of thioflavin T binding to amyloid fibrils. J Struct Biol 151:229–238
Klunk WE (1998) Biological markers of Alzheimer’s disease. Neurobiol Aging 19:145–147
Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Mathis CA (2001) Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci 69:1471–1484
Krebs MR, Bromley EH, Donald AM (2005) The binding of thioflavin-T to amyloid fibrils: localisation and implications. J Struct Biol 149:30–37
Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT Jr (2002) Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature 418:291
Mathis CA, Bacskai BJ, Kajdasz ST, McLellan ME, Frosch MP, Hyman BT, Holt DP, Wang Y, Huang GF, Debnath ML, Klunk WE (2002) A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett 12:295–298
Mirzabekov TA, Lin MC, Kagan BL (1996) Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem 271:1988–1992
Murphy RM (2007) Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim Biophys Acta 1768:1923–1934
Nesterov EE, Skoch J, Hyman BT, Klunk WE, Bacskai BJ, Swager TM (2005) In vivo optical imaging of amyloid aggregates in brain: design of fluorescent markers. Angew Chem Int Ed Engl 44:5452–5456
Nilsson MR (2004) Techniques to study amyloid fibril formation in vitro. Methods 34:151–160
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60:759–767
Ono K, Yamada M (2006) Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J Neurochem 97:105–115
Pramanik D, Dey SG (2011) Active site environment of heme-bound amyloid beta peptide associated with Alzheimer’s disease. J Am Chem Soc 133:81–87
Pratico D, Lee VMY, Trojanowski JQ, Rokach J, Fitzgerald GA (1998) Increased F2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo. FASEB J 12:1777–1783
Robinson SR, Bishop GM (2002) A beta as a bioflocculant: implications for the amyloid hypothesis of Alzheimer’s disease. Neurobiol Aging 23:1051–1072
Scaria PV, Atreyi M, Rao MVR (1986) Helix-beta conformational transition of poly(l-lysine) on dye binding. Biopolymers 25:2349–2358
Sumner Makin O, Serpell LC (2004) Structural characterisation of islet amyloid polypeptide fibrils. J Mol Biol 335:1279–1288
Takuma K, Yan SS, Stern DM, Yamada K (2005) Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer’s disease. J Pharmacol Sci 97:312–316
Turnbull S, Tabner BJ, Brown DR, Allsop D (2003) Generation of hydrogen peroxide from mutant forms of the prion protein fragment PrP121-231. Biochemistry 42:7675–7681
Volkova KD, Kovalska VB, Balanda AO, Vermeij RJ, Subramaniam V, Slominskii YL, Yarmoluk SM (2007) Cyanine dye-protein interactions: looking for fluorescent probes for amyloid structures. J Biochem Biophys Methods 70:727–733
Yuan C, Yi L, Yang Z, Deng Q, Huang Y, Li H, Gao Z (2012) Amyloid beta-heme peroxidase promoted protein nitrotyrosination: relevance to widespread protein nitration in Alzheimer’s disease. J Biol Inorg Chem 17:197–207
Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA (2001) Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem 76:435–441
Zhu X, Raina AK, Lee HG, Casadesus G, Smith MA, Perry G (2004) Oxidative stress signalling in Alzheimer’s disease. Brain Res 1000:32–39
Acknowledgments
The authors appreciate the joint financial support of this investigation by the research councils of the Kermanshah University of Medical Sciences and Razi University, Kermanshah, Iran.
Conflict of interest
The authors do not declare any conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ghadami, S.A., Hossein-pour, Z., Khodarahmi, R. et al. Synthesis and in vitro characterization of some benzothiazole- and benzofuranone-derivatives for quantification of fibrillar aggregates and inhibition of amyloid-mediated peroxidase activity. Med Chem Res 22, 115–126 (2013). https://doi.org/10.1007/s00044-012-0012-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-012-0012-3