
Abstract
We have described a new method to heterogenize s-triazine catalyst by easy preparation of ionic liquid-based s-triazine-immobilized silica-coated Fe3O4 magnetic nanoparticles [Fe3O4@SiO2-s-triazinium chloride]. The structure of the newly prepared nanoparticles was characterized by Fourier transform infrared, X-ray powder diffraction, scanning electron microscopy, transmission electron microscopy, energy dispersive X-ray method, vibrating sample magnetometery and thermo-gravimetric analysis. These nanoparticles were identified as an efficient catalyst for one-pot three-component synthesis of 4,8-dihydropyrano[3,2-b]pyran derivatives in excellent yields. The catalyst was easily separated by magnetic decantation, and the recovered nanoparticles were reused for four fresh runs without any significant loss of catalytic activity. The synthesized dihydropyrano[3,2-b]pyrans were evaluated for their antioxidant and antifungal activities and found they are relatively active.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, the enhanced environmental concerns have prompted the development and application of ionic liquids (ILs) as alternative reaction media and/or as task specific catalysts in various organic syntheses [1,2,3,4,5]. The research interest having been developed on ionic liquids is mainly because of their favorable properties such as intrinsic ion conductivity, non-volatility, high thermal stability, negligible vapor pressure, tunable acidity, and selective dissolvability [6,7,8,9]. Despite these promising advantages, ILs are still impeded in their widespread practical applications as catalysts or reaction media for several drawbacks including: (1) high viscosity limiting the catalytic power of the ILs, (2) homogeneous nature of ILs making the product separation and catalyst recovery difficult, and (3) high cost due to the use of relatively large amounts of ILs [10,11,12]. The most promising alternative solution to overcome these problems is the development of IL-based heterogeneous catalysts [13,14,15]. In this context, the strategy of immobilizing ILs onto solid supports such as carbon nanotubes,[16,17,18] graphene oxide [19, 20], mesoporous silica [21,22,23], and for the preparation of various IL-based heterogeneous catalysts, amorphous silica [24, 25], has recently been deployed. The aforementioned physical and chemical features of ILs are not retained in these catalysts, but they offer the high performance of recycling and easy separation making them practically suitable in continuous industrial processes [26].
Recently, magnetic nanoparticles (MNPs), especially magnetic nano-metal oxides [27], have found wide applications as excellent supports for various catalysts and ILs [28,29,30,31,32,33], because of their high surface-area that provides high catalyst loading capacity, magnetic susceptibility, catalytic activity, and long-term stability [34,35,36]. Moreover, these nonmagnetic catalysts can be easily separated and recycled using an external magnet which prevents from the tedious process of isolation and cyclization via filtration or centrifugation [29, 37, 38].
Multi-component reactions (MCRs) have emerged as efficient tools in the sustainable synthesis of organic and biologically active compounds including heterocyclic products [39,40,41]. In organic synthesis, the MCRs have recently received considerable interest because they offer versatile and powerful strategy featured by high-synthetic efficiency and atom-economical assembling of structurally divers and complex molecules [42].
4H-Fused pyrans such as 2-amino-4H-pyrano[3,2-b]pyran-carbonitrile derivatives belong to an important class of biologically active heterocyclic compounds [43,44,45]. These compounds have been shown to exhibit a wide range of biological and pharmaceutical properties such as antimicrobial [46], antifungal [47], anti-HIV [48], anticancer [49], calcium channel antagonist activity [50], antimalarial [51], anti-diabetic [52], and anti-inflammatory activities [53]. The methods recently reported for the synthesis of 2-amino-4H-pyrano[3,2-b]pyran-carbonitrile scaffolds are mainly based on one-pot three-component reactions [46,47,48, 54]. However, most of these methods suffer from certain drawbacks such as low yields, high temperature, high cost of reagents and catalysts, and longer reaction times. Therefore, the development of more convenient and efficient new methods for preparation of these compounds appears to be most challenging. In the last few years, 5-hydroxy-2-hydroxymethyl-4H-pyran-4-one (kojic acid) has found considerable interest as easily available and biologically active precursor in the three-component reactions with malononitrile and aldehydes for the preparation of pharmaceutically important dihydropyrano[3,2-b]pyran-3-carbonitriles [55, 56].
As a continuation of our interest in developing environmentally benign and efficient methodologies for the synthesis of various heterocyclic compounds and other organic transformations [57,58,59,60,61], we report, herein, the synthesis of a new type of magnetically separable s-triazine-based ionic liquid immobilized onto silica-coated Fe3O4 nanoparticles. The synthesized Fe3O4@SiO2-s-triazinium chloride MNPs were successfully examined as heterogeneous recyclable catalyst for the synthesis of 2-amino-4H-pyrano[3,2-b]pyran-carbonitrile scaffolds from one-pot three-component reactions between aryl aldehydes, malononitrile and 5-hydroxy-2-hydroxymethyl-4H-pyran-4-one (kojic acid) in water (Scheme 1).

Synthesis of 4H-pyrano[3,2-b]pyran-carbonitrilies catalyzed by Fe3O4@SiO2-s-triazinium chloride MNPs
In the following stage of the present research, we were encouraged to evaluate the biological activities of the synthesized 2-amino-4H-pyrano[3,2-b]pyran-carbonitriles 4a–j as potential antioxidants using 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging assay as reported by Mensor et al. [62]. Moreover, the antifungal activity of the synthesized products was assessed against Fusarium oxysporum bacteria and the corresponding inhibitory zone values were determined.
Experimental details
General
Melting points were measured in open capillary tubes using a BUCHI 510 apparatus. Fourier transform infrared (FT-IR) spectra were recorded from KBr pellets on a Perkin Elmer GX FT-IR spectrometer. 1H NMR and 13C NMR spectra were recorded on 250 and 400 MHz BRUKER AVANCE instruments at 400 MHz and 100 MHz, respectively, for samples in DSMO-d6 as a solvent at ambient temperature using tetramethylsilane (TMS) as internal standard. Scanning electron microscopy (SEM) images were obtained on an EM3200 instrument operated at 30 kV accelerating voltage. Energy-dispersive X-ray (EDX) analysis was carried out using a FESEM-SIGM (German) instrument. The curves obtained from thermo-gravimetric analysis (TGA) were recorded in air using TGA/DTA PYRIS DIAMOND. Magnetic measurement of the catalyst was performed using a vibrating sample magnetometer (VSM) instrument MDKFT. In addition, high resolution transmission electron microscopy (TEM) was conducted on the nanoparticles using a HRTEM Philips CM30, (300 kV) instrument.
Synthesis of IL-based s-triazine-functionalized Fe3O4@SiO2 magnetic nanoparticles
The sequential synthesis of the IL-based s-triazine-immobilized silica-coated Fe3O4 nanoparticles is illustrated in Scheme 2 as explained below.

Sequential preparation of IL-based Fe3O4@SiO2-s-triazinium chloride MNPs
Preparation of magnetic Fe3O4 nanoparticles
The Fe3O4 nanoparticles were prepared by co-precipitation according to the previously reported procedure [63]. In a typical procedure, a mixture of FeCl3·6H2O (5.84 g) and FeCl2·4H2O (2.15 g) was dissolved in double distilled water (60 mL). Then, to the mixture was quickly added 25% NH4OH solution (15 mL) and the mixture was vigorously stirred at 80 °C for 4 h. The precipitated Fe3O4 nanoparticles were separated with an external magnet and washed with distilled water and ethanol three times until the pH value reached the neutral position. Then, the prepared nanoparticles were dried in a vacuum oven at 60 °C.
Modification of the magnetic Fe3O4 nanoparticles
The silica-modification of the prepared Fe3O4 nanoparticles was performed using the well-known Stӧber’s method with little modification [64]. First, the as-prepared Fe3O4 nanoparticles (1 g) were dispersed in mixed ethanol (100 mL) and deionized water (10 mL) containing 2.5 mL 25% ammonia solution. To the rapidly stirred dispersion, was added tetraethyl orthosilicate (TEOS) (2 mL) and stirring was continued for 2 h at 60 °C. The resulted Fe3O4@SiO2 magnetic nanoparticles were separated magnetically, washed repeatedly with deionized water and ethanol, and then dried in air for 4 h.
Synthesis of 3-chloropropyl-bonded Fe3O4@SiO2 nanoparticles (Fe3O4@SiO2–(CH2)3–Cl)
The Fe3O4@SiO2 nanoparticles (1 g) were added to a solution of 3-(chloropropyl)triethoxysilane (CPTES) (3 mL) in dried toluene (100 mL) followed by stirring at 60 °C for 4 h under nitrogen atmosphere. The precipitated chloropropyl-bonded magnetic nanoparticles Fe3O4@SiO2–(CH2)3–Cl were separated by a magnet, washed with toluene, and dried under vacuum.
Immobilization of s-triazine onto the Fe3O4@SiO2–(CH2)3–Cl nanoparticles
A suspension of the above-prepared Fe3O4@SiO2–(CH2)3–Cl nanoparticles (0.5 g) in toluene (70 mL) was placed in an ultrasonic bath and irradiated at room temperature for 20 min. Then, to the resulted mixture was added s-triazine (2 mmol) followed by stirring at 90 °C for 6 h under N2 atmosphere. Eventually, the mixture was cooled to room temperature to precipitate the s-triazine-functionalized nanoparticles Fe3O4@SiO2-s-triazinium chloride which were separated by an external magnet, washed with ethanol, and dried under vacuum at room temperature.
General procedure for synthesis of 4H-dihydropyrano[3,2-b]pyran-3-carbonitrile derivatives
To the mixture of aromatic aldehyde (1 mmol), malononitrile (1 mmol), 5-hydroxy-2-hydroxymethyl-4H-pyran-4-one (kojic acid) (1 mmol) in H2O (5 mL), was added Fe3O4@SiO2-s-triazinium chloride MNPs (0.02 g), and the resulted mixture was refluxed at 100 °C for an appropriate time (Table 4). After completion of the reaction as monitored by TLC, the catalyst was magnetically separated using an external magnet, dried under vacuum, and recrystallized from hot ethanol to obtain the pure products 4a–k (Table 4).
Characterization data
2-Amino-4-(2,6-dichlorophenyl)-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (4c)
White solid; mp. 249–251 °C; IR (KBr, cm−1): 3313, 3273 (NH2), 3431 (OH), 2195 (CN), 1644 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 4.07–4.13 (ABq, 1H, 2J = 16 Hz, CHaliph), 4.15–4.21 (ABq, 2J = 16 Hz, 1H, CHaliph), 5.65–5.69 (t,1H, OH), 5.83 (s, 1H, CHvinyl), 6.35 (s, 1H, CHaliph), 7.33 (s, 2H, NH2), 7.38–7.59 (m, 3H, CHarom) ppm; 13C NMR (400 MHz, DMSO-d6): 40.1, 52.2, 59.5, 111.6, 118.6, 129.0, 130.6, 132.8, 135.2, 137.6, 145.6, 160.0, 168.1, 169.2 ppm; MS: m/z = 364 (M+).
2-Amino-6-(hydroxymethyl)-4-(4-nitrophenyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (4d)
Dark brown solid; mp. 243–245 °C; IR (KBr, cm−1): 3337, 3451 (NH2), 3326 (OH), 2194 (CN), 1649 (C=O), 190, 1351 (NO2); 1H NMR (400 MHz, DMSO-d6): δ 4.09–4.13 (ABq, 2J = 16 Hz, 1H, CHaliph), 4.17–4.21 (ABq, 2J = 16 Hz, 1H,CHaliph), 5.07 (s, 1H, CHvinyl), 5.69 (s, 1H, OH), 6.35 (s, 1H, CHaliph), 7.38 (s, 2H, NH2), 7.60–7.63 (m, 4H, CHarom), 8.24–8.26 (m, 2H, CHarom) ppm; 13C NMR (400 MHz, DMSO-d6): δ 40.1, 54.6, 59.0, 110.5, 118.9, 124.1, 129.3, 136.7, 147.1, 147.7, 147.9, 159.3, 168.3, 169.5 ppm; MS: m/z = 341 (M+).
2-Amino-6-hydroxymethyl-4-(4-hydroxyphenyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (4 g)
Cream solid; mp. 240–242 °C; IR (KBr, cm−1): 3423, 3351 (NH2), 3233 (OH), 2200 (CN), 1657 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 4.09–4.15 (ABq, 2J = 16 Hz, 1H, CHaliph), 4.18–4.23 (ABq, 2J = 16 Hz, 1H, CHaliph), 4.64 (s, 1H, CHvinyl), 5.67–5.70 (t, 1H, OH), 6.31 (s, 1H, CHaliph), 7.16 (s, 2H, NH2), 6.67–7.07 (m, 4H, CHarom), 9.49 (s,1H, OH) ppm; 13C NMR (400 MHz, DMSO-d6): δ 56.0, 59.1, 111.2, 115.5, 115.6, 119.4, 128.8, 131.1, 136.0, 149.5, 157.0, 159.1, 168.1, 169.6 ppm; MS: m/z = 312 (M+).
2-Amino-6-hydroxymethyl-4-(naphthalen-1-yl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (4 h)
Light purple solid; mp. 252–254 °C; IR (KBr, cm−1): 3290, 3173 (NH2), 3395 (OH), 2204 (CN), 1668 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 3.89–3.95 (ABq, 2J = 16 Hz, 1H, CHaliph), 4.08–4.12 (ABq, 2J = 16 Hz, 1H, CHaliph), 5.61–5.64 (t, 1H, OH), 5.76 (s, 1H, CHvinyl), 6.33 (s, 1H, CHaliph), 7.22 (s, 2H, NH2), 7.26–8.35 (m, 7H, CHarom) ppm; 13C NMR (400 MHz, DMSO-d6): δ 40.1, 56.1, 59.0, 111.4, 119.3, 123.2, 126.0, 126.1, 126.6, 126.8, 128.5, 128.8, 131.1, 133.6, 136.7, 137.0, 149.8, 159.5, 168.3, 169.6 ppm; MS: m/z = 346 (M+).
2-Amino-6-hydroxymethyl-4-(naphthalen-2-yl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile (4i)
Yellow solid; mp. 250–252 °C; IR (KBr, cm−1): 3385, 3190 (NH2), 3302 (OH), 2190 (CN), 1642 (C=O); 1H NMR (400 MHz, DMSO-d6): δ 4.05–4.11 (ABq, 2J = 8 Hz, 1H, CHaliph), 4.15–4.21 (ABq, 2J = 16 Hz, 1H, CHaliph), 4.98 (s, 1H, CHvinyl), 5.64–5.67 (t, 1H, OH), 6.34 (s, 1H, CHaliph), 7.85 (s, 2H, NH2), 7.90–7.97 (m, 7H, CHarom) ppm; 13C NMR (400 MHz, DMSO-d6): δ 40.6, 55.8, 58.9, 59.1, 111.4, 119.3, 125.5, 126.3, 126.5, 126.7, 127.6, 127.9, 128.9, 132.7, 136.5, 138.3, 148.9, 159.2, 168.3, 169.6 ppm; MS: m/z = 346 (M+).
Biological evaluation
Antioxidant activity
The radical scavenging activities of the synthesized compounds 4a–j were evaluated against the stable DPPH radical based on the method reported by Mensor et al. [62]. The stock solutions (2.5 mL) of the samples at different concentrations (0.2–1 mg mL−1) in methanol was prepared. Then, 1.0 mL of each compound was mixed with 1 mL of 0.3 mM methanol solution of DPPH radical with vigorously shaking. Then, the samples were incubated in the dark at room temperature for 30 min. The absorbance was observed against a blank solution at 517 nm using the UV/V spectrophotometer. The assay was carried out in triplicate using ascorbic acid as a standard, and the antioxidant activity, namely the percentage of inhibition, was calculated according to the following formula (Table 4),
in which, As is the absorbance of the reaction mixture containing samples and DPPH, Ab stands for the absorbance of the mixture containing samples + methanol, and Ac represents the absorbance of the control sample containing DPPH + methanol. Additionally, representing the sample concentration causing 50% inhibition, IC50 value was determined (Fig. 8).
Antifungal activity
In vitro antifungal activities of the compounds 4a–j were screened against Fusarium oxysporum cultivated in Potatoes Dextrose Agar (PDA) medium. The samples were added to the sterilized cultivation medium at a concentration of 200 ppm. To make the control groups, double distilled water and DMSO were added to the plates. After a 7-day incubation at the temperature 27 °C of fungus on culture medium containing samples, radial growth of fungal mycelium was recorded. The inhibition rate (%) was calculated using the following formula,
where R is the radial growth of fungal mycelia on the control plate and r is the radial growth of fungal mycelia on the plate treated with the new derivatives. All the resulting data summarized in Table 5 are the average of triplicate assays.
Statistical analysis
Statistical analysis of variance was performed using Student’s t test by SPSS program and p value < 0.05 was regarded to be significant. Data are expressed as means ± standard deviation.
Results and discussion
Synthesis and characterization of the catalyst Fe3O4@SiO2-s-triazinium chloride
To prepare the Fe3O4@SiO2-supported s-triazinium chloride MNPs was initially prepared by the co-precipitation of ferrous (Fe2+) and ferric (Fe3+) ions in basic aqueous solution of NH4OH under reflux condition in deionized water according to the reported procedure [61]. Then, the prepared Fe3O4 nanoparticles were coated with the silica layer on their surface using the well-known Stӧber method [64], by treatment of Fe3O4 suspension in alkaline ethanol/water solution with tetraethyl orthosilicate (TEOS). In the next step, the chloropropyl group as a linker was bonded to the silicon atom to prepare the Fe3O4@SiO2–(CH2)3–Cl nanoparticles by treatment of a suspension of Fe3O4@SiO2 nanoparticles in toluene with a solution of 3-(chloropropyl)triethoxysilane (CPTES) in dried toluene. Finally, the immobilization of s-triazine moiety onto the surface of Fe3O4@SiO2–(CH2)3–Cl nanoparticles was achieved by the addition of s-triazine to the suspension of the Fe3O4@SiO2–(CH2)3–Cl nanoparticles in toluene under ultrasonic irradiation condition at room temperature to obtain the Fe3O4@SiO2-s-triazinium chloride MNPs (Scheme 2).
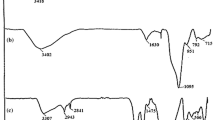
The FT-IR spectra of the bare Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2–(CH2)3–Cl, and Fe3O4@SiO2-s-triazinium chloride MNPs shown in Fig. 1 clearly display the characteristic bands around 3422, 1078 and 574 cm−1 ascribed to the ν(O–H), ν(Si–O), and ν(Fe–O) characteristic bands, respectively. The appearance of the stretching bands around 2345 and 1078 cm−1 due to the Si–O group indicated that the nanoparticles were successfully coated with silica groups on their surface. In addition, the stretching bands shown at 2800–3000 cm−1 in the IR spectra of Fe3O4@SiO2–(CH2)3–Cl (1c) and Fe3O4@SiO2-s-triazinium chloride (1d) nanoparticles are assigned to the aliphatic CH2 group confirming the successful loading of chloropropyl linker onto the magnetic nanoparticles. Added to this, the successful immobilization of s-triazine moiety onto the surface of Fe3O4@SiO2–(CH2)3–Cl nanoparticles is clearly verified by the appearance of the bands at 1728–1590 cm−1 attributed to the C–N stretching band of the s-triazine group (Fig. 1d).

FT-IR spectra of a Fe3O4, b Fe3O4@SiO2, c Fe3O4@SiO2–(CH2)3–Cl and d Fe3O4@SiO2–(CH2)3-s-triazinium chloride MNPs
In Fig. 2, the X-ray powder diffraction patterns (XRD) of Fe3O4 and Fe3O4@SiO2-s-triazinium chloride MNPs are shown. The crystal planes at 2θ = 19.37°, 30.53°, 35.69°, 43.28°, 57.20°, 53.85°, 62.93° corresponding to their Miller indices (111, 220, 311, 400, 422, 511, 440) respectively, with including the diffraction peaks typical for amorphous silica in the range 2θ = 20–30°, are related to the pure crystalline Fe3O4 nanoparticles. As shown in Fig. 2b, the same diffraction peaks are present in the XRD pattern obtained for the Fe3O4@SiO2-s-triazinium chloride nanoparticles. These diffraction peaks and indexed planes confirm excellent crystallinity of these nanoparticles. The diffraction peaks of pure Fe3O4 nanoparticles match well with highly crystalline cubic spinel structure (JCPDS no. 65-3107) [65]. These results provided the evidence that the crystal structure of pure Fe3O4 did not change after immobilization with s-triazine moiety. The crystal size of the Fe3O4@SiO2-s-triazinium chloride can be determined from the XRD pattern using Debye–Scherrer’sequation, which gives the relationship between the peak broadening and the particle size. The peak width, size and inter planar distance from the XRD pattern of supported nanoparticles were investigated and found to be ranging from 30.5° to 62.9° with the achieved results summarized in Table 1. The average core diameter of the nanoparticles calculated from the XRD results using the aforementioned equation was found to be 32 nm.

XRD Diffraction patterns of a bare Fe3O4 and b Fe3O4@SiO2-s-triazinium chloride MNPs
The morphological structure and particle distribution of the Fe3O4@SiO2-s-triazinium chloride MNPs were analyzed by scanning electron microscopy (SEM) analysis. In agreement with the SEM image presented in Fig. 3a, the encapsulated particles of Fe3O4@SiO2-s-triazinium chloride are well dispersed and exhibit approximately spherical morphology made up of nano-sized particles with an average diameter of 34 nm. In supporting analysis by transmission electron microscopy (TEM) conducted on the synthesized Fe3O4@SiO2-s-triazinium chloride nanoparticles (Fig. 3b), it has been demonstrated that, these nanoparticles are quasi-spherical in shape possessing almost a uniform distribution of particle size. However, as inferred from the presence of dark spots inside the bright spherical silica thin shell, these magnetic nanoparticles can be considered to have core–shell structure.

a SEM and b TEM images of Fe3O4@SiO2-s-triazinium chloride MNPs
The chemical identification of the elements contained in the catalyst Fe3O4@SiO2-s-triazinium chloride was determined by energy dispersive X-ray (EDX) analysis. The EDX spectral pattern shown in Fig. 4, clearly indicated the chemical characterization of the typical catalyst sample with the peaks for C, N, O, Si, Fe, Cl elements, confirming the formation and dispersion of Fe3O4@SiO2-s-triazinium chloride nanoparticles. Therefore, these results justify the successful grafting of s-triazinium moiety onto the surface of Fe3O4@SiO2 MNPs linked by propyl group.

EDX spectrum of Fe3O4@SiO2-s-triazinium chloride MNPs (presence of signals for Au is allocated to the sample grid)
The magnetic properties of Fe3O4 and Fe3O4@SiO2-s-triazinium chloride MNPs were studied by vibrating sample magnetometer (VSM) at 300 K with the field sweeping range from − 9000 to + 9000 Oersted (Fig. 5). According to the magnetization curves shown in Fig. 5, these nanoparticles perform reasonably high super paramagnetic properties. The magnetization saturation (Ms) values of the bare Fe3O4 and Fe3O4@SiO2-s-triazinium MNPs were found to be 56 and 38 emu g−1 respectively. The decrement in the Ms value of Fe3O4@SiO2-s-triazinium chloride in comparison to the Ms value of the bare Fe3O4 nanoparticles can be related to the decrease in the particle size due to the silica layer coated on the surface of the particles, and also due to the functionalization with triazine moiety. Despite this reduction in Ms value of Fe3O4@SiO2-s-triazinium chloride nanoparticles, they still have considerable paramagnetic property to be magnetically separable from reaction mixture.

VSM patterns of a uncoated Fe3O4 and b Fe3O4@SiO2-s-triazinium chloride MNPs
The thermal gravimetric analysis (TGA) was performed on Fe3O4@SiO2-s-triazinium chloride nanoparticles to obtain information about their thermal stability and behavior. According to the TGA pattern shown in Fig. 6, the stepwise decreasing in the weight percentage of the nanoparticles take place in three consecutive steps from about 100 to 750 °C. It is likely that, the first weight loss of ca. 3% at about 140 °C is due to the removal of physically adsorbed solvents and moisture. The second weight loss of about 12% occurring within the range 200–500 °C (centered at 430 °C) possibly corresponds to decomposition of organic species and triazine group grafted to the surface of the nanoparticles. In the final step, the complete decomposition of nanoparticles happens beyond 500 °C. These results clearly verify the successful grafting of triazine moiety to the silica-coated Fe3O4 nanoparticles.

TGA diagram of Fe3O4@SiO2-s-triazinium chloride MNPs
Catalytic activity of Fe3O4@SiO2-s-triazinium chloride MNPs
The catalytic activity of the prepared Fe3O4@SiO2-s-triazinium chloride MNPs was examined as heterogeneous recyclable ionic liquid-based magnetic nanocatalyst in three-component synthesis of 4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile derivatives. In the initial step, to screen the reaction parameters, the three-component condensation reaction between benzaldehyde, malononitrile, and 5-hydroxy-2-hydroxymethyl-4H-pyran-4-one (kojic acid) was chosen as model reaction (Table 2). First, we studied the effect of various solvents such as water, ethanol, and acetonitrile on the reaction in the presence of Fe3O4@SiO2-s-triazinium chloride (0.01 g) at room temperature. The best result was observed when the reaction was performed using water as the solvent in which the hydrogen bonding prevails the solvent polarity (entry 1). Next, the temperature effect was studied by conducting further experiments at 60 and 100 °C in water as the solvent of choice and the optimal temperature was found to be 100 °C (entry 6). In further studies, regarding the effect of catalyst loading, the optimal amount of the catalyst was achieved to be 0.02 g and the use of higher amounts of catalyst (entry 10) did not have any improving effect on the yield. The indispensable use of the catalyst Fe3O4@SiO2-s-triazinium chloride in the reaction was substantiated by conducting the reaction in the absence of the catalyst which resulted in no detectable amount of the respective product (entry 12).

To establish the scope and generality of the reaction, a divers series of aldehydes 4a–k bearing different substituents were reacted in the presence of Fe3O4@SiO2-s-triazinium chloride catalyst under the aforementioned optimized conditions (Scheme 1). The experimental results summarized in Table 3 clearly revealed that, all the reactions proceeded smoothly and the relevant products 4a–k were produced in excellent yields (85–98%) and relatively short reaction times (20–45 min). In addition, the heteroaromatic aldehydes 1j and 1k were also conveniently reacted under the optimal condition to yield the corresponding products 4j and 4k in excellent yields (entries 10, 11). As seen in Table 3, the aldehydes containing electron-withdrawing groups generally display higher reactivity in comparison to the aldehydes carrying electron-releasing groups. The products were characterized on the basis of their physical and spectral (FT-IR, 1H NMR, 13C NMR, MS) data and compared with the reported data in the case of the known products.
Proposed reaction mechanism
A plausible mechanism similar to the previously reported mechanism for other basic catalysts in the same reaction [56, 66], has been suggested to describe the formation of 2-amino-4-aryl-6-(hydroxymethyl)-8-oxo-4,8-dihydropyrano[3,2-b]pyran-3-carbonitriles from one-pot three-component reaction between aromatic aldehydes, malononitrile, and kojic acid under the catalytic effect of IL-based Fe3O4@SiO2-s-triazinium chloride MNPs as depicted in Scheme 3. Initially, deprotonation of malononitrile is affected by the basic triazinium moiety of the catalyst to generate the corresponding anion which undergoes dehydrative addition to the aldehyde to furnish 2-arylidenemalononitrile intermediate (I). Similarly, the removal of proton from 5-hydroxy group of kojic acid under the same catalytic effect results in the formation of the enolate ion (II). Subsequently, nucleophilic addition of the enolate ion (II) to the intermediate (I) takes place to form the adduct (III) followed by consecutive intramolecular cyclization and tautomerization to yield the expected products.

A possible mechanism for the synthesis of dihydropyrano[3,2-b]pyran-3-carbonitriles catalyzed by Fe3O4@SiO2-s-triazinium chloride MNPs
Catalyst recyclability
The recyclability and reusability potentials of the catalyst Fe3O4@SiO2-s-triazinium chloride were examined for the model reaction. After the reaction completion, the catalyst was recovered from the reaction mixture simply using an external magnet. Then, the separated solid was washed with hot ethanol and dried at room temperature. The separated catalyst was reused under the same optimized conditions for four consecutive fresh runs without any noticeable drop in the yield of the product and catalytic activity. The slight decrease in the yield of the product can be attributed to the normal loss of the product and catalyst during the work-up process (Fig. 7).

Recyclability test of Fe3O4@SiO2-s-triazinium chloride MNPs
Biological assessment
Antioxidant activity
Antioxidants are recognized as an important class of biologically active natural products and synthetic compounds playing a key role in reducing the risk of chronic diseases such as cancer and other diseases [68]. Antioxidants are characterized by their high biological performance as scavengers, reducers and quenching agents to prevent the biological systems from the free radical damages [69,70,71]. In the last few decades, there has been a great deal of attention towards the production of new types of antioxidant agents without posing any harmful side effects. 2,2-Diphenyl-1-picrylhydrazyl (DPPH) is a free radical, showing hydrogen acceptor ability towards antioxidants. Hence, it is commonly used in DPPH assay for measuring the antioxidant activity of different natural and synthetic samples [72]. With regard to the promising biological properties of pyrano[3,2-b]pyran derivatives, we were encouraged to study the free radical scavenging activity of the synthesized dihydropyrano[3,2-b]pyran-3-carbonitriles 4a–j (C1–C10) using the DPPH assay. According to the experimental results summarized in Table 4, and graphical presentation in Fig. 8, many of these compounds perform relatively high antiradical properties (16.40–81.54%) in comparison with ascorbic acid as a standard antiradical agent (80.16%). In general, the higher antioxidant activity is reflected in a lower IC50 value. Ascorbic acid, also known as vitamin C, has been used as a standard with an IC50 value of 0.127–0.006 mg mL−1 in the present study exhibiting a lower IC50 value than most of the compounds which are assessed in the present study (IC50; 0.96 ± 0.09 to 2.43 ± 0.07 mg mL−1) (Fig. 8). As shown in Fig. 8, the radical scavenging efficiencies of the compounds C1–C10 against DPPH radicals range in descending order as: ascorbic acid > 4d > 4c > 4e > 4f > 4b > 4j > 4h > 4i˃4a˃4g.

Comparison between antioxidant activities (IC50 values) of the samples (C1–C10) and that of the ascorbic acid as a standard antioxidant agent
Experiments were performed in triplicate and expressed as mean ± SD. Values in each row with different superscripts are significantly different (p ≤ 0.05).
Antifungal activity
It is a well-known fact that, many types of fungal bacteria species such as Fusarium species are hosted by different kinds of plants and agricultural crops throughout the world that cause soil-borne diseases of great economic concern. An area of media, where bacteria are unable to grow, owing to the presence of a drug which impedes their growth, is called inhibition zone. Fusarium oxysporum (F. oxysporum) is one of the soil-borne pathogens that causes serious damages to the plants and harvests of great economic importance [73, 74]. Here, the potential antifungal activities of the synthesized products were assessed and their corresponding inhibitory zone values were assessed against F. oxysporum bacteria (Table 5). Accordingly, it was observed that the solvent dimethyl sulfoxide (DMSO) used as negative control exhibited no activity against F. oxysporum, while the test samples performed excellent antifungal activities (49–90%) as shown in Table 5. Among these samples, the samples C1 (4e), C8 (4i) and C10 (4a) performed highest antifungal activities in this system at a concentration of 200 ppm. Usually, most of the old synthetic fungicides are resistant to environmental degradation through chemical, biological, and photolytic processes and have human health impacts. Therefore, our tested samples can be conveniently used as alternative antifungal drugs to increase the resistance of a wide variety of crop plants.
Experiments were carried out in triplicate and expressed as mean ± SD. Values shown in the row with different superscripts are significantly different (p ≤ 0.05).
Conclusion
In summary, we have successfully immobilized, for the first time, s-triazine ring onto the magnetic Fe3O4@SiO2 nanoparticles to prepare the s-triazinium-based ionic liquid Fe3O4@SiO2-s-triazinium chloride magnetic nanoparticles. The structure of the synthesized catalyst was fully characterized by FT-IR, EDX, SEM, XRD, TGA, and VSM analytical techniques. This newly synthesized nanocatalyst has been explored as an efficient, versatile and recyclable heterogeneous nanocatalyst in three-component synthesis of dihydropyrano[3,2-b]pyran-3-carbonitrile derivatives in water in excellent yields. The catalyst can be easily recovered simply by an external magnet and reused four consecutive times with no significant loss of activity. Next, the synthesized products were biologically assessed and found to possess reasonably good antioxidant and antifungal activities.
References
M.J. Earle, K.R. Seddon, Pure Appl. Chem. 72, 1391 (2000)
S.-G. Lee, Chem. Commun. 10, 1049 (2006)
J.H. Davis, James Jr., Chem. Lett. 33, 1072 (2004)
C. Paun, J. Barklie, P. Goodrich, H. Gunaratne, A. McKeown, V. Parvulescu, C. Hardacre, J. Mol. Catal. A Chem. 269, 64 (2007)
B.C. Ranu, S. Banerjee, Org. Lett. 7, 3049 (2005)
M.A. Zolfigol, S. Baghery, A.R. Moosavi-Zare, S.M. Vahdat, H. Alinezhad, M. Norouzi, RSC Adv. 4, 57662 (2014)
H. Alinezhad, M. Tajbakhsh, M. Norouzi, S. Baghery, World Appl. Sci. J. 22, 1711 (2013)
A.C. Cole, J.L. Jensen, I. Ntai, K.L. Tran, K.J. Weaver, D.C. Forbes, J.H. Davis, J. Am. Chem. Soc. 124, 5962 (2002)
T. Welton, Coord. Chem. Rev. 248, 2459 (2004)
J. Miao, H. Wan, G. Guan, Cat. Commun. 12, 353 (2011)
J. Safari, Z. Zarnegar, Monatsh. Chem. 144, 1389 (2013)
S. Sahoo, P. Kumar, F. Lefebvre, S.B. Halligudi, Appl. Catal. A 354, 17 (2009)
M.H. Valkenberg, C. deCastro, W.F. Hölderich, Green Chem. 4, 88 (2002)
M.H. Valkenberg, C. de Castro, W.F. Hölderich, Top. Catal. 14, 139 (2001)
H. A.Corma, A. Garcı´a, Leyva, Tetrahedron 60, 8553 (2004)
X. Wu, F. Zhao, J.R. Varcoe, A.E. Thumser, C. Avignone-Rossa, R.C. Slade, Bioelectrochemistry 77, 64 (2009)
Z. Zarnegar, J. Safari, New J. Chem. 40, 7986 (2016)
L. Rodríguez-Pérez, E. Teuma, A. Falqui, M. Gómez, P. Serp, Chem. Commun. 35, 4201 (2008)
W.-H. Zhang, P.-P. He, S. Wu, J. Xu, Y. Li, G. Zhang, X.-Y. Wei, Appl. Catal. A Gen. 509, 111 (2016)
W. Zheng, R. Tan, S. Yin, Y. Zhang, G. Zhao, Y. Chen, D. Yin, Catal. Sci. Technol. 5, 2092 (2015)
W. Xie, L. Hu, X. Yang, Ind. Eng. Chem. Res. 54, 1505 (2015)
B. Zou, Y. Hu, L. Jiang, R. Jia, H. Huang, Ind. Eng. Chem. Res. 52, 2844 (2013)
B. Zhen, Q. Jiao, Y. Zhang, Q. Wu, H. Li, Appl. Catal. A Gen. 445, 239 (2012)
Y. Gu, C. Ogawa, S. Kobayashi, Org. Lett. 9, 175 (2007)
H. Hagiwara, Y. Sugawara, K. Isobe, T. Hoshi, T. Suzuki, Org. Lett. 6, 2325 (2004)
C. Yuan, Z. Huang, J. Chen, Catal. Commun. 24, 56 (2012)
P. Wang, H. Liu, J. Niu, R. Li, J. Ma, Catal. Sci. Technol. 4, 1333 (2014)
M.B. Gawande, A.K. Rathi, P.S. Branco, R.S. Varma, Appl. Sci. 3, 656 (2013)
R.B. Nasir Baig, R.S. Varma, Chem. Commun. 49, 752 (2013)
Y. Zhu, L.P. Stubbs, F. Ho, R. Liu, C.P. Ship, J.A. Maguire, N.S. Hosmane, Chem. Catal. Chem. 2, 365 (2010)
R. Tayebee, M. Amini, H. Rostamian, A. Aliakbari, Dalton Trans. 43, 1550 (2014)
R. Tayebee, M. Amini, N. Abdollahi, A. Aliakbari, S. Rabiee, H. Ramshini, Appl. Catal. A Gen. 468, 75 (2013)
V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrera, J.M. Basset, Chem. Rev. 111, 3036 (2011)
G.B. Dharma, M.P. Kaushik, A.K. Halve, Tetrahedron Lett. 53, 2741 (2012)
L. Ma’mani, M. Sheykhan, A. Heydari, M. Faraji, Y. Yamini, Appl. Catal. A Gen. 377, 64 (2010)
X. Zheng, S. Liu, L. Zhang, J.P. Cheng, Green Chem. 11, 455 (2009)
Q.M. Kainz, O. Reiser, Acc. Chem. Res. 47, 667 (2014)
D. Guin, B. Baruwati, S.V. Manorama, Org. Lett. 9, 1419 (2007)
B. Jiang, T. Rajale, W. Wever, S.J. Tu, G. Li, Chem. Asian. J. 5, 2318 (2010)
Y. Zou, Y. Hu, H. Liu, D. Shi, ACS Comb. Sci. 14, 38 (2012)
Z. Huang, Y. Hu, Y. Zhou, D. Shi, ACS Comb. Sci. 13, 45 (2011)
C. de Graaff, E. Ruijter, R.V.A. Orru, Chem. Soc. Rev. 41, 3969 (2012)
L. Bonsignore, G. Loy, D. Secci, A. Calignano, Eur. J. Med. Chem. 28, 517 (1993)
W. Kemnitzer, S. Jiang, Y. Wang, S. Kasibhatla, C. Crogan-Grundy, M. Bubenik, D. Labrecque, R. Denis, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng, J. Drewe, S.X. Cai Bioorg. Med. Chem. Lett. 18, 603 (2008)
N. Foloppe, L.M. Fisher, R. Howes, A. Potter, A.G.S. Robertson, A.E. Surgenor, Bioorg. Med. Chem. 14, 4792 (2006)
L. Moafi, S. Ahadi, A. Bazgir, Tetrahedron Lett. 51, 6270 (2010)
B.V. Subba Reddy, M. Ramana Reddy, G. Narasimhulu, J.S. Yadav, Tetrahedron Lett. 51, 5677 (2010)
S. Gao, C.H. Tsai, C. Tseng, C.-F. Yao, Tetrahedron 64, 9143 (2008)
M.P. Zhu, I. Kimiaki, Tetrahedron Lett. 38, 5301 (1997)
X. Xiong, M.C. Pirrung, Org. Lett. 10, 1151 (2008)
M. Sefkow, H. Kaatz, Tetrahedron Lett. 40, 6561 (1999)
R.C. Fox, P.D. Taylor, Synth. Commun. 28, 1575 (1998)
J. Safari, S.H. Banitaba, S. Dehghan Khalili, J. Mol. Catal. A Chem. 335, 46 (2011)
D. Ding, C.-G. Zhao, Tetrahedron Lett. 51, 1322 (2010)
S.H. Banitaba, J. Safari, S. Dehghan, Khalili, Ultrason. Sonochem. 20, 401 (2013)
B. Sadeghi, P. Farokhi, S. Hashemian, J. Chem. Res. 38, 54 (2014)
A. Azarifar, R. Nejat-Yami, M. AlKobaisi, D. Azarifar, J. Iran.Chem. Soc. 10, 439 (2013)
D. Azarifar, F. Soleimanei, RSC Adv. 4, 12119 (2014)
D. Azarifar, O. Badalkhani, Y. Abbasi, J. Sulf. Chem. 37, 656 (2016)
D. Azarifar, M. Ghaemi, Appl. Organometal. Chem. 36, 1981 (2017)
D. Azarifar, Y. Abbasi, Synth. Commun. 46, 745 (2016)
L. Mensor, F.S. Menezes, G.G. Leitao, A.S. Reis, T.S. Santos, C.S. Coube, Phytother. Res. 15, 127 (2001)
M. Ma, Y. Zhang, W. Yu, H.Y. Shen, H.Q. Zhang, N. Gu, Colloids Surf. A 212, 219 (2003)
W. Stober, A. Fink, E.J. Bohn, J. Colloid Interface Sci. 26, 62 (1968)
T.Z. Yang, C.M. Shen, H.J. Gao, J. Phys. Chem. B 109, 23233 (2005)
X.X. Menga, B.X. Dua, B. Zhaoa, Y.L. Lia, C.F. Chenb, J. Chem. Res. 78, 638 (2013)
J. Mondal, A. Modak, A. Bhaumik, J. Mol. Catal. A Chem. 335, 236 (2011)
R.M. Seabra, P.B. Andrade, P. Valentao, E. Fernandes, F. Carvalho, M.L. Bastos. In: Fingerman, M., Nagabhushanam, R. (eds.) Biomaterials from Aquatic and Terrestrial Organisms, vol 5, p. 115. Science Publishers, Enfield (2006)
L. Yu, J. Perret, B. Davy, J. Wilson, C.L. Melby, Food Chem. Toxicol. 67, 2600 (2002)
R.L. Prior, X.M. Wu, K. Schaich, J. Agric. Food Chem. 53, 4290 (2005)
A.J. Parr, J.P. Bolwell, J. Sci. Food Agric. 80, 985 (2002)
D. Villano, M.S. Fernandez-Pachon, M.L. Moya, A.M. Troncoso, M.C. Garcia-Parilla, Talanta 71, 230 (2007)
G.N. Agrios, Plant Pathology, 3rd ed, vol 803. Academic Press, Inc., New York (1988)
P.R. Larkin, D.R. Fravel. Plant Dis. 82, 1022 (1998)
Acknowledgements
The authors wish to thank the Research Council of Bu-Ali Sina University for financial support to carry out this research. Also, Mr. Jamshid Rakhtshah from Tabriz University is greatly acknowledged for his generous contribution of some chemical materials utilized in this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Azarifar, D., Ebrahimiasl, H., Karamian, R. et al. s-Triazinium-based ionic liquid immobilized on silica-coated Fe3O4 magnetic nanoparticles: an efficient and magnetically separable heterogeneous catalyst for synthesis of 2-amino-4,8-dihydropyrano[3,2-b]pyran-3-carbonitrile derivatives for antioxidant and antifungal evaluation studies. J IRAN CHEM SOC 16, 341–354 (2019). https://doi.org/10.1007/s13738-018-1521-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-018-1521-x