
Abstract
The ionic liquid 1-methyl-3-(3-trimethoxysilylpropyl)imidazolium chloride was immobilized on Fe3O4 nanoparticles and used as an efficient and reusable catalyst for the one-pot synthesis of 1,2,4,5-tetrasubstituted imidazoles at room temperature under ultrasonic irradiation. The immobilized ionic liquid catalysts proved to be effective and easily separated from the reaction media by applying an external magnetic field. This procedure has many obvious advantages compared to those reported in the previous literatures, including avoiding the use of harmful catalysts, reacting at room temperature, high yields, and simplicity of the methodology.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the past decade, many investigations have been conducted to try to develop greener chemical processes and synthetic methods because of environmental considerations. Several new methods and novel-designed materials have been developed to make the chemical reactions cleaner and more benign. For example, performing safer and environmentally friendly synthetic processes is now possible by using ionic liquids [1, 2].
In recent years, ionic liquids have attracted increasing interest in the context of green synthesis [3–6]. Although ionic liquids were initially introduced as alternative green reaction media because of their unique chemical and physical properties of nonvolatility, nonflammability, thermal stability, and controlled miscibility [4–6], nowadays they have evolved beyond this, showing their significant role in controlling reactions as catalysts [7, 8].
Though ionic liquids possessed such promising advantages, their widespread practical application was still hampered by several drawbacks: (1) high viscosity, which resulted in only a minor part of ionic liquids taking part in the catalyzed reaction, (2) homogeneous reaction, which was difficult for product separation and catalyst recovery, and (3) consequently high costs for the use of relatively large amounts of ionic liquids [9, 10]. Therefore, in order to solve these problems mentioned earlier, immobilized ionic liquid catalyst combining the advantageous characteristics of ionic liquids, inorganic acids, and solid acids were proposed [11, 12].
On the other hand, magnetic nanoparticles (MNPs) have received a great deal of attention because of their potential biomedical applications in fields such as drug delivery [13, 14], magnetic resonance imaging [15], biomolecular sensors [16], bioseparation [17], and magneto-thermal therapy [18]. Additionally, recent studies show that magnetic nanoparticles are excellent supports for catalysts [19]. The supported catalysts proved to be effective and easily separated from the reaction media by applying an external magnetic field.
Imidazole derivatives are a very interesting class of heterocyclic compounds because they have many pharmacological properties and play important roles in biochemical processes [20, 21]. In recent years, the synthesis of 2,4,5-trisubstituted imidazoles has been performed by catalysts such as Yb(OPf)3 [22], ZrCl4 [23], NiCl2·6H2O/Al2O3 [24], silica sulfuric acid (SSA) [25], polymer-supported ZnCl2 [26], and phosphomolybdic acid [27]. Also some catalysts used for 1,2,4,5-tetrasubstituted imidazoles including silica gel or zeolite InCl3·3H2O [28], silica gel/NaHSO4 [29], HClO4–SiO2 [30], heteropolyacids [31], BF3·SiO2 [32], FeCl3·6H2O [33], and alumina [34] are applied as common catalysts for 2,4,5-trisubstituted and 1,2,4,5-tetrasubstituted imidazoles.
Despite their potential utility, some of these methods are not environmentally friendly and suffer from one or more disadvantages, for example, hazardous reaction conditions, complex workup and purification, strongly acidic conditions, high temperature, use of toxic metal catalysts, poor yields, occurrence of side reactions, and long reaction time. Therefore, the development of clean, high-yielding, and environmentally friendly approaches is still a challenge for organic chemists in the synthesis of highly substituted imidazoles [21].
Ionic liquids (ILs) have been used as efficient catalysts for an improved and rapid synthesis of imidazoles [35–37]. Zang et al. [38] synthesized 2-aryl-4,5-diphenyl imidazoles using ionic liquid 1-ethyl-3-methylimidazole acetate as catalyst at room temperature under ultrasonic irradiation. The ionic liquid N-methyl-2-pyrrolidonium hydrogen sulfate has been used as an efficient and reusable catalyst for the one-pot synthesis of 2,4,5-trisubstituted and 1,2,4,5-tetrasubstituted imidazoles under thermal solvent-free conditions in excellent yields [39].
During the last 3 decades, ultrasound-accelerated organic chemical reactions have been increasingly developed by researchers across the globe for the synthesis of organic molecules. Ultrasound irradiation offers an alternative energy source for organic reactions that are ordinarily accomplished by heating. Ultrasound-assisted reactions proceed by the formation, growth, and collapse of acoustic bubbles in the reaction medium. These directly help in shortening the time span of reactions and increasing the yield of products [40].
In this work, our objective is to functionalize magnetite nanoparticles with 1-methyl-3-(3-trimethoxysilylpropyl)imidazolium chloride to obtain a heterogeneous catalyst for the synthesis of 1,2,4,5-tetrasubstituted imidazoles. To the best of our knowledge, there are no reports on the synthesis of highly substituted imidazoles catalyzed by magnetic nanoparticle-supported ionic liquid under ultrasonic irradiation. The heterogeneous catalyst could be recovered easily and reused many times without significant loss of its catalytic activity (Scheme 1).

Results and discussion
Characterization of the prepared IL-MNPs
Scheme 2 shows the sequence of events in the functionalization of MNPs with 1-methyl -3-(3-trimethoxysilylpropyl)-1H-imidazol-3-ium chloride. In the first step, the magnetite nanoparticles of 18–20 nm were prepared by coprecipitation of iron(II) and iron(III) ions in basic solution at 85 °C using the method described by Massart [43]. Then, 1-methyl-3-(3-trimethoxysilylpropyl)imidazolium chloride (IL) was prepared from the reaction of N-methylimidazole with (3-chloropropyl)trimethoxysilane at 80 °C. In the second step, the external surface of MNPs was coated with IL to obtain IL-MNPs (Scheme 2).

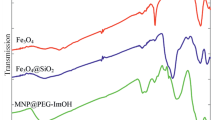
Figure 1 shows the Fourier transform infrared (FTIR) spectra of both the unfunctionalized and functionalized magnetic nanoparticles. The Fe–O stretching vibration near 580 cm−1, O–H stretching vibration near 3,432 cm−1, and O–H deformed vibration near 1,625 cm−1 were observed for both as shown in Fig. 1a, b. The significant features observed in Fig. 1b are the appearance of the peaks at 1,007 cm−1 (Si–O stretching) and at 2,800 cm−1 (–CH2 stretching). These results provided the evidence that IL-methyl was successfully attached to the surface of Fe3O4 nanoparticles.

The comparative FT-IR spectra for a MNPs and b IL-MNPs
Figure 2 presents the XRD diffraction patterns of the prepared MNPs and IL-MNPs. The position and relative intensities of all peaks conform well with the standard XRD pattern of Fe3O4 (JCPDS card no. 79-0417), indicating retention of the crystalline cubic spinel structure during functionalization of MNPs. The XRD patterns of the particles show eight characteristic peaks and reveal a cubic iron oxide phrase (2θ = 30.35, 35.95, 43.45, 53.70, 57.25, 62.88, 71.37, 74.46°). These are related to their corresponding indices (2 2 0), (3 1 1), (4 0 0), (3 3 1), (4 2 2), (3 3 3), (4 4 0), and (5 3 1), respectively [35]. It is implied that the resultant nanoparticles are pure Fe3O4 with a spinel structure and that the grafting process did not induce any phase change of Fe3O4.

XRD patterns of a MNPs, b IL-MNPs
The crystal size of MNP and IL-MNP nanoparticles can be determined from the XRD pattern using Debye-Scherrer’s equation.
where D(h k l) is the average crystalline diameter, 0.94 is the Scherrer’s constant, λ is the X-ray wavelength, β is the half width of XRD diffraction lens, and θ is the Bragg’s angle in degrees. Here, the (3 1 1) peak of the highest intensity was picked out to evaluate the particle diameter of the nanoparticles. MNPs and IL-MNPs were calculated to be 12 nm and 30 nm, respectively.
The IL-functionalized magnetic Fe3O4 nanoparticles could be separated to the sidewall of the container after 30 s using a magnet of 2,000 G, suggesting the obtained magnetic nanoparticles had an excellent magnetic responsivity, which prevents composite nanoparticles from aggregation and enables them to redisperse rapidly when the magnetic field is removed.
The SEM image in Fig. 3a shows that magnetite Fe3O4 nanoparticles have a mean diameter of about 18 nm and a nearly spherical shape. Figure 3b shows that IL-MNP particles still keep the morphological properties of Fe3O4 except for a slightly larger particle size and smoother surface (more than 20 nm in size), where silica is uniformly coated on the Fe3O4 particles to form a silica shell.

The SEM images of a MNPs and b IL-MNPs
The magnetization curve for Fe3O4 nanoparticles and IL-MNPs is shown in Fig. 4. Room temperature-specific magnetization (M) versus applied magnetic field (H) curve measurements of the sample indicates a saturation magnetization value (M s ) of 40.5 emu g−1, lower than that of bare magnetic nanoparticles (60.1 emu g−1) because of the coated shell. In Fig. 4 we can also see that the two magnetization curves both follow a Langevin behavior over the applied magnetic field and the coercivity (H C) could be ignored, which can be considered as superparamagnetism [44].

Magnetization curves for the prepared MNPs and IL-MNPs at room temperature
Evaluation of the catalytic activity of IL/MNPs through the synthesis of 1,2,4,5-tetrasubstituted imidazoles
Initially, we sought a green, facile, and efficient method for the synthesis of 1,2,4,5-tetrasubstituted imidazoles catalyzed by IL-MNPs under ultrasonic irradiation at room temperature (Scheme 1). Our investigation began with the evaluation of IL-MNPs as a catalyst in the reaction of benzil (1 mmol), benzaldehyde (1 mmol), aniline (1 mmol), and ammonium acetate (5 mmol) at 40 kHz under sonication. The use of 0.15 g of IL-MNPs in ethanol afforded an 87 % yield (Table 1, entry 1) of the desired product. Optimization of the reaction conditions was undertaken to increase the yield employing catalyst loadings in a wide variety of solvents. The results were summarized in Table 1. The yield increased to 94 % using 0.1 g of IL-MNPs (Table 1, entry 2). However, a reduction in yield was observed by decreasing the catalyst loading to 0.05 g (Table 1, entry 3). In the absence of the catalyst, the reaction proceeded sluggishly (Table 1, entry 4). Obviously, the catalyst is an essential component of the reaction.
The investigation of reaction medium for the process revealed that reaction solvents played a significant role in the model reaction. We examined different solvents such as water, EtOH, THF, CH3CN, and CH2Cl2 on a model reaction under ultrasound irradiation at room temperature (Table 1, entries 5–8). Furthermore, the effect of ultrasound irradiation on the reaction was also studied. The model reaction was carried out with stirring at room temperature. The reaction failed to give low yield under high-speed stirring conditions (Table 1, entry 9). It was apparent that the ultrasound irradiation accelerates this transformation under ambient conditions. The reason may be the phenomenon of cavitation produced by ultrasound [45]. Hence, the conditions of Table 1, entry 2 were the optimized reaction conditions.
To show the merit of the present work in comparison with reported results in the literature, we compared results of IL-MNPs with reported catalysts in the synthesis of 1,2,4,5-tetrasubstituted imidazoles. As shown in Tables 1 and 2, the immobilized ionic liquid can act as a suitable catalyst with respect to reaction times and yields of the products.
The substrate scope of the reaction was then evaluated using a variety of structurally diverse aldehydes and amines. The presence of electron withdrawing groups on the aromatic aldehydes and primary amines produced high yields of 1,2,4,5-tetrasubstituted imidazoles.
Most of the products are known and were identified by comparison of their physical and spectral data with those of authentic samples. All melting points compared satisfactorily with those reported in the literature.
The possibility of recycling the catalyst was examined using the reaction of benzil, benzaldehyde, aniline, and ammonium acetate under optimized conditions. Upon completion, the catalyst was separated by an external magnet and was washed with acetone, and the recycled catalyst was saved for the next reaction. The recycled catalyst could be reused six times without any further treatment. No observation of any appreciable loss in the catalytic activity of nanocatalyst was observed (Fig. 5). As observed in Fig. 6, the XRD of the recovered magnetite nanocatalyst was indexed according to the magnetite phase (JCPDS card no. 79-0417), and so there is no considerable change in its magnetic phase. Therefore, the heterogeneous nanocatalyst is stable during synthesis of tetrasubstituted imidazoles.

Recyclability of IL-MNPs in the reaction of benzil (1 mmol), benzaldehyde (1 mmol), aniline (1 mmol), and ammonium acetate (5 mmol) under ultrasonic waves (40 kHz) at room temperature

XRD patterns of recovered IL-MNPs after four recoveries
In summary, we have been able to introduce an efficient and environmentally friendly approach for the synthesis of biologically active tetrasubstituted imidazoles by one-pot four-component condensation of benzil, aldehydes, amines, and ammonium acetate using IL-MNPs as a recyclable catalyst under ultrasonication at room temperature. Corrosiveness, safety, less waste, ease of separation, and recovery are all among desirable factors for the chemical industry, which we have considered in our green chemistry approach. The approach has several benefits, for example, low waste, easy workup, short reaction time, and high yields.
Experimental
Chemical reagents in high purity were purchased from Merck Chemical Co. All materials were of commercial reagent grade. Melting points were determined in open capillaries using an Electrothermal Mk3 apparatus. 1H NMR and 13C NMR spectra were recorded with a Bruker DRX-400 spectrometer at 400 and 100 MHz, respectively. NMR spectra were obtained in DMSO-d 6 and CDCl3 solutions and are reported as parts per million (ppm) downfield from tetramethylsilane as internal standard. The abbreviations used are: singlet (s), doublet (d), triplet (t), and multiplet (m). FT-IR spectra were obtained with potassium bromide pellets in the range 400–4,000 cm−1 with a Perkin-Elmer 550 spectrometer. Mass spectrum was recorded by a QP-1100EX Shimadzu spectrometer. The element analyses (C, H, N) were obtained from a Carlo ERBA Model EA 1108 analyzer carried out on a Perkin-Elmer 240c analyzer. The UV–vis measurements were obtained with a GBC cintra 6 UV–vis spectrophotometer. Nanostructures were characterized using a Holland Philips Xpert X-ray powder diffraction (XRD) diffractometer (CuK, radiation, λ = 0.154056 nm) at a scanning speed of 2°/min from 2θ = 10° to 100°. Scanning electron microscopy (SEM) was performed on a FEI Quanta 200 SEM operated at a 20-kV accelerating voltage. The samples for SEM were prepared by spreading a small drop containing nanoparticles onto a silicon wafer and being dried almost completely in air at room temperature for 2 h; they then were transferred onto SEM conductive tapes. The transferred sample was coated with a thin layer of gold before measurement. Sonication was performed in a UP 400S ultrasonic processor equipped with a 3-mm-wide and 140-mm-long probe, which was immersed directly into the reaction mixture. The operating frequency was 40 kHz, and the output power was 0–400 W using manual adjustment.
Preparation of the magnetic Fe3O4 nanoparticles (MNPs)
Fe3O4–MNPs were prepared using chemical coprecipitation as described in the literature [41] with little modification. Typically, 20 mmol of FeCl3·6H2O and 10 mmol of FeCl2·4H2O were dissolved in 75 cm3 of distilled water in a three-necked round-bottom flask (250 cm3) under Ar atmosphere for 1 h. Thereafter, under rapid mechanical stirring, 10 cm3 of NaOH (10 M) was added to the solution within 30 min with vigorous mechanical stirring and ultrasound treatment. After being rapidly stirred for 1 h, the resultant black dispersion was heated to 85 °C for 1 h. The black precipitate formed was isolated by magnetic decantation, exhaustively washed with double-distilled water, then ethanol, and dried at 60 °C in vacuum.
1-Methyl-3-(3-trimethoxysilylpropyl)-1H-imidazol-3-ium chloride (IL)
1-Methylimidazole (13.6 cm3, 0.17 mol) and 31 cm3 (3-chloropropyl)trimethoxysilane (0.17 mol) were refluxed at 80 °C for 3 days in the absence of any catalyst and solvent under Ar atmosphere. The unreacted materials were washed by diethyl ether (3 × 8 cm3). The diethyl ether was removed under reduced pressure at room temperature, followed by heating under high vacuum, to yield a yellowish viscous liquid. Isolated yield was 98 %. The product was identified through the spectroscopic data, which were confirmed by comparison with those reported in the literature [42].
Modification of magnetic nanoparticles with IL to obtain IL-MNPs
Freshly prepared magnetite nanoparticles (2 g) were suspended in 250 cm3 ethanol (95 %) and sonicated for 60 min. The resulting suspension was mechanically stirred, followed by addition of a solution of 100 cm3 ethanol (95 %) containing 6 g IL (18.5 mmol) and 1 cm3 concentrated ammonia (28 %). Stirring under Ar was continued for 36 h. The modified magnetite nanoparticles were magnetically separated and washed three times with 50 cm3 ethanol (95 %) and then dissolved in 200 cm3 methanol and stirred mechanically for 30 min. Ether (50 cm3) was added and the modified nanoparticles were magnetically separated, washed with 50 cm3 ether, and dried under vacuum for 24 h. Typically, 3–3.5 g of brownish black powder could be obtained.
General procedure for the synthesis of 1,2,4,5-tetrasubstituted imidazoles
To a solution of benzil (1 mmol), aldehyde (1 mmol), 0.4 g ammonium acetate (5 mmol), and primary aliphatic or aromatic amine (1 mmol) in 10 cm3 ethanol, 0.1 g MNPs-IL was added and the reaction mixture was exposed to ultrasonic irradiation at room temperature. The progress of the reaction was followed by TLC. After the reaction had been completed, the catalyst was separated by an external magnet, and the reaction mixture was dissolved in acetone and filtered. The filtrate was concentrated on a rotary evaporator under reduced pressure and the solid product obtained was washed with water and recrystallized from acetone–water (9:1, v/v). Pure products were obtained in excellent yields, as summarized in Table 2.
2-(3,4-Dimethoxyphenyl)-1,4,5-triphenyl-1H-imidazole (5j, C29H24N2O2)
M.p.: 178–180 °C; UV–vis (EtOH): λ max = 311 nm; IR (KBr): \( \bar{\nu } \) = 3,045, 1,617, 1,578 cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 3.61 (s, J = 8.4 Hz, 6H), 6.85 (d, J = 8.8 Hz, 2H), 7.15–7.33 (m, 16H) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 55.9, 112.7, 115.6, 121.2, 122.0, 122.3, 123.8, 126.9, 128.7, 130.1, 130.5, 132.5, 132.8, 150.4, 152.8, 153.3 ppm; MS (70 eV): m/z (%) = 432 (M+, 55), 417 (50), 402 (44), 77 (32).
2,4,5-Triphenyl-1-propyl-1H-imidazole (5 k, C24H22N2)
M.p.: 87–89 °C; UV–vis (EtOH): λ max = 284 nm; IR (KBr): \( \bar{\nu } \) = 3,025, 1,597, 1,479 cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 0.51 (t, J = 6.8 Hz, 3H), 1.32 (m, J = 6.8 Hz, 2H), 3.81 (t, J = 7.2 Hz, 2H), 7.10–7.55 (m, 13H), 7.7 (d, J = 6.8 Hz, 2H) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 11.5, 24.0, 53.6, 121.5, 122.8, 124.0, 125.2, 126.2, 126.9, 128.4, 128.8, 130.1, 132.8, 156.7 ppm; MS (70 eV): m/z (%) = 338 (M+, 68), 323 (65), 309 (57), 295 (43), 15 (40).
2-(4-Chlorophenyl)-4,5-diphenyl-1-propyl-1H-imidazole (5l, C24H21N2Cl)
M.p.: 85–87 °C; UV–vis (EtOH): λ max = 294 nm; IR (KBr): \( \bar{\nu } \) = 3,025, 1,645, 1,489 cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 0.51 (t, J = 6.8 Hz, 3H), 1.34 (m, J = 6.8 Hz, 2H), 3.81 (t, J = 7.2 Hz, 2H), 7.15-7.30 (m, 12H), 7.34 (d, J = 7.2 Hz, 2H) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 11.5, 24.2, 53.5, 121.5, 122.7, 124.3, 125.8, 126.2, 127.0, 128.4, 129.1, 130.4, 132.8, 135.6, 156.6 ppm; MS (70 eV): m/z (%) = 435 (M+, 55), 432 (53), 417 (50), 402 (44), 77 (32).
2-(4-Methylphenyl)-4,5-diphenyl-1-propyl-1H-imidazole (5m, C25H24N2)
M.p.: 78–83 °C; UV–vis (EtOH): λ max = 294 nm; IR (KBr): \( \bar{\nu } \) = 3,028, 1,620, 1,497 cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 0.52 (t, J = 6.8 Hz, 3H), 1.30 (m, J = 6.8 Hz, 2H), 2.50 (s, 3H), 3.80 (t, J = 7.2 Hz, 2H), 7.12-7.35 (m, 12H), 7.50 (d, J = 8 Hz, 2H) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 11.3, 22.0, 24.2, 53.4, 121.1, 122.9, 124.0, 125.3. 126.2, 127.0, 127.9, 128.8, 130.2, 130.9, 133.0, 142.4, 156.2 ppm; MS (70 eV): m/z (%) = 352 (M+, 70), 323 (52), 309 (59), 295 (46), 15 (37).
2-(4-Methoxyphenyl)-4,5-diphenyl-1-propyl-1H-imidazole (5n, C25H24N2O)
M.p.: 76–80 °C; UV–vis (EtOH): λ max = 305 nm; IR (KBr): \( \bar{\nu } \) = 3,016, 1,628, 1,510 cm−1; 1H NMR (400 MHz, DMSO-d 6): δ = 0.50 (t, J = 6.8 Hz, 3H), 1.33 (m, J = 6.8 Hz, 2H), 3.05 (s, 3H), 3.81 (t, J = 7.2 Hz, 2H), 7.10-7.30 (m, 12H), 7.46 (d, J = 8 Hz, 2H) ppm; 13C NMR (100 MHz, DMSO-d 6): δ = 11,5, 24.3, 53.4, 55.9, 117.1, 121.2, 122.8, 123.8, 125.3, 126.2, 126.8, 128.7, 130.1, 132.0, 132.6, 156.7, 160.8 ppm; MS (70 eV): m/z (%) = 368 (M+, 59), 337 (52), 323 (59), 309 (48), 234 (46), 31 (30).
References
Roger RD, Seddon KR, Volkov S (eds) (2003) Green industrial applications of ionic liquids. Kluwer Academic Publishers, Dordrecht
Abu-Reziq R, Wang D, Post M, Alper H (2008) Chem Mater 20:2544
Burrell AK, Sesto RED, Baker SN, McCleskey TM, Baker GA (2007) Green Chem 5:449
Sheldon R (2001) Chem Commun 23:2399
Wilkes JS (2002) Green Chem 4:73
Jain N, Kumar A, Chauhan S, Chauhan SMS (2005) Tetrahedron 61:1015
Zang H, Wang M, Cheng BW, Song J (2009) Ultrason Sonochem 16:301
Borikar SP, Daniel T (2011) Ultrason Sonochem 18:928
Sahoo S, Kumar P, Lefebvre F, Halligudi SB (2009) Appl Catal A 354:17
Zhu LL, Liu YH, Chen J (2009) Ind Eng Chem Res 48:3261
Chen W, Zhang YY, Zhu LB, Lan JB, Xie RG, You JS (2007) J Am Chem Soc 129:13879
Miao J, Wan H, Guan G (2011) Catal Commun 12:353
Zhu Y, Fang Y, Kaskel S (2010) J Phys Chem C 114:16382
Neuberger T, Schoepf B, Hofmann H, Hofmann M, Rechenberg B (2005) J Magn Magn Mater 293:483
Pankhurst QA, Connolly J, Jones SK, Dobson J (2003) J Phys D Appl Phys 36:167
Graham DL, Ferreira HA, Freitas PP (2004) Trends Biotechnol 22:455
Wang D, He J, Rosenzweig N, Rosenzweig Z (2004) Nano Lett 4:409
Jordan A, Scholz R, Wust P, Fahling H, Felix R (1999) J Magn Magn Mater 201:413
Hu A, Yee GT, Lin W (2005) J Am Chem Soc 127:12486
Puratchikody A, Doble M (2007) Bioorg Med Chem Lett 15:1083
Safari J, Khalili SD, Rezaei M, Banitaba SH, Meshkani F (2010) Monatsh Chem 141:1339
Shen M, Cai C, Yi W (2008) J Fluorine Chem 129:541
Sharma GVM, Jyothi Y, Sree Lakshmi P (2006) Synth Commun 36:2991
Heravi MM, Bakhtiari K, Oskooie HA, Taheri S (2007) J Mol Catal A: Chem 263:279
Shaabani A, Rahmati A, Farhangi E, Badri Z (2007) Catal Commun 8:1149
Wang L, Cai C (2009) Monatsh Chem 140:541
Jadhave SD, Kokare ND, Jadhave SD (2009) J Heterocycl Chem 45:1461
Sharma SD, Hazarika P, Konwar D (2008) Tetrahedron Lett 49:2216
Karimi AR, Alimohammadi Z, Azizian J, Mohammadi AA, Mohammadzadehi MR (2006) Catal Commun 7:728
Kantevari S, Vuppalapati SVN, Biradar DO, Nagarapu L (2007) J Mol Catal A: Chem 266:109
Heravi MM, Derikvand F, Bamoharram FF (2007) J Mol Catal A: Chem 263:112
Sadeghi B, Mirjalili BBF, Hashemi MM (2008) Tetrahedron Lett 4:2575
Heravi MM, Derikv F, Haghighi M (2008) Monatsh Chem 139:31
Usyatinsky AY, Khmelnitsky YL (2000) Tetrahedron Lett 41:5031
Siddiqui SA, Narkhede UC, Palimkar SS, Daniel T, Lahoti RJ, Srinivasan KV (2005) Tetrahedron 61:3539
Chary MV, Keerthysri NC, Vupallapati S, Lingaiah N, Kantevari S (2008) Catal Commun 9:2013
Xia M, Lu Y (2007) J Mol Catal A: Chem 265:205
Zang H, Su Q, Mo Y, Cheng BW, Jun S (2010) Ultrason Sonochem 17:749
Shaterian HR, Ranjbar M (2011) J Mol Liquids 160:40
Nagargoje D, Mandhane P, Shingote S, Badadhe P, Gill C (2012) Ultrason Sonochem 19:94
Yang D, Hu J, Fu S (2009) J Phys Chem C 113:7646
Chrobok A, Baj S, Pudło W, Jarzebski A (2009) Appl Catal A: General 366:22
Massart R (1981) IEEE Trans Magn 17:1247
Jiang Y, Jiang J, Gao Q, Ruan M, Yu H, Qi L (2008) Nanotech 19:75714
Kappe CO (2004) Angew Chem Int Ed 43:6250
Kidwai M, Mothsra P, Bansal V, Somvanshi RK, Ethayathulla AS, Dey S, Singh TP (2007) J Mol Catal A: Chem 265:177
Khosropour AR (2008) Ultrason Sonochem 15:659
Murthy SN, Madhav B, Nageswar YVD (2010) Tetrahedron Lett 51:5252
Shelke KF, Sapkal SB, Sonar SS, Madje BR, Shingate BB, Shingare MS (2009) Bull Korean Chem Soc 30:1057
Acknowledgments
We gratefully acknowledge the financial support from the Research Council of the University of Kashan for supporting this work by Grant No. 256722/I.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Safari, J., Zarnegar, Z. Magnetic nanoparticle supported ionic liquid as novel and effective heterogeneous catalyst for synthesis of substituted imidazoles under ultrasonic irradiation. Monatsh Chem 144, 1389–1396 (2013). https://doi.org/10.1007/s00706-013-1015-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-013-1015-6