
Abstract
5-Hydroxypentylammonium acetate as a task-specific ionic liquid promotes efficient tandem Knoevenagel-phospha-Michael reaction of phosphite esters with aryl/heteroaryl/alkyl/salicylaldehydes and malonitrile/ethyl cyanoacetate at room temperature in short reaction times. This simple procedure allows a series of β-phosphonomalonates and 4-substituted 2-amino-4H-chromenes with phosphonic acid dialkyl esters to be synthesized in good to high yields in the presence of an ionic liquid for the first time.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phosphonates are fascinating and versatile compounds in organic synthesis. They have unique properties which expand their applications as enzyme inhibitors, metabolic probes, peptide mimetic, antibiotics, and pharmacological agents besides to their traditional roles as intermediates in organic synthesis [1–7]. For example, phosphonoacetic acid [8–11] has been shown to inhibit the replication of cytomegalovirus and herpes virus by interacting directly with the virus-induced DNA polymerase. Phosphonoformate [12] has shown activity in cell cultures against HTLV-III (the virus implicated in AIDS). A derivative of β-phosphonomalonic acid [13] was used as an inhibitor of ras farnesyl protein transferase in studies directed toward the development of new antitumor agents.
Extensive efforts have been made to introduce convenient and efficient methods for the synthesis of phosphonates. Direct P–C bond formation represents one of the most versatile and powerful tools for the synthesis of phosphonates. Amongst the methods for P–C bond formation, phospha-Michael addition, that is, the addition of a phosphorous nucleophile to an electron-deficient alkene has evoked remarkable attention by organic chemists [14–28]. Synthesis of β-phosphonomalonates by this method is most commonly promoted by BrØnsted/Lewis acids [14–16], bases [17–22], transition metals [23, 24], radical initiators such as AIBN [25, 26] or microwaves [27]. Although these methods are valuable, they suffer from one or more of the following drawbacks such as: low yields; high temperature; long reaction times; requiring a promoter such as microwave; using toxic solvents or a large amount of catalyst and tedious work-up procedures. Therefore, the development of a new method to overcome these shortcomings still remains an ongoing challenge for the synthesis of these significant scaffolds.
Low melting point salts consisting of ions which are often classified as ionic liquids exist in the liquid state at ambient temperatures. In recent years, room temperature ionic liquids have received considerable attention as an alternative green reaction medium for numerous organic reactions due to their favorable properties, such as good solvating capability, wide liquid range, negligible vapor pressure, tunable polarity, high thermal stability and ease of recyclability [29–31]. They also played a significant role in controlling the reactions as catalysts [32–34].
As part of our continued interest on the synthesis of phosphonate derivatives [35–46], we have recently concentrated on the development of new procedures for the synthesis of β-phosphonomalonates [40–46]. Herein, in view of the projected benefits of ionic liquids in organic transformations, we wish to introduce an eco-friendly method for the efficient synthesis of β-phosphonomalonates directly from aldehydes, malononitrile and trialkyl phosphites via tandem Knoevenagel-phospha-Michael reaction in the presence of 5-hydroxypentylammonium acetate (5-HPAA) [47–54] as a reusable ionic liquid at room temperature. This procedure is the first report on the synthesis of β-phosphonomalonates in an ionic liquid.
Experimental
Chemicals were purchased from Merck and Fluka Chemical Companies. Known compounds were easily identified by comparison of their spectroscopic data with those reported [19, 55–60]. The unknown compounds were properly characterized by their spectroscopic (IR, 1H NMR, 13C NMR, MS and elemental analysis) data. Melting points were determined by Buchi 510 apparatus and are uncorrected. IR spectra were run on a Perkin–Elmer 780 instrument. NMR spectra were recorded on a Bruker Avance DPX-250 and 400. Mass spectra were recorded on a Shimadzu GCMS-QP5050A. Elemental analysis for C, H and N was obtained using a Elementar, Vario EL III. The purity of the products and the progress of the reactions were accomplished by TLC on silica-gel polygram SILG/UV254 plates.
A typical procedure for the synthesis of 5-HPAA
5-HPAA was prepared according to the previously reported procedure for the synthesis of 2-hydroxyethylammonium acetate [49]. To a stirring solution of 5-amino-pentan-1-ol (50 mmol, 5.15 g) in EtOH (15 mL), a solution of acetic acid (0.5 mmol, 3 g) in EtOH (15 mL) was added dropwise at room temperature within 1 h. The resultant solution was stirred at room temperature for another 20 h. EtOH was removed in vacuo and the oil in residue was dried in vacuo at 50 °C for 48 h to give 5-HPAA as a light yellow viscous liquid. 1H NMR (400 MHz, DMSO-d 6 ): δ 1.32–1.37 (m, 2 H), 1.39–1.42 (m, 2 H), 1.48–1.54 (m, 2 H), 1.69 (s, 3 H), 2.67 (t, 2 H, 3 J HH = 7.6 Hz), 3.38 (t, 2 H, 3 J HH = 6.4 Hz), 6.78 (s, 4 H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ 22.9, 25.0, 28.1, 32.4, 39.4, 60.8, 175.7 ppm; MS (EI, 70 eV), m/e (%): 146 (3 M+–NH3 + or M+–OH), 104 (100 M+–−OAc), 59 (18 OAc−).
General procedure for the one-pot synthesis of β-phosphonomalonates in the presence of 5-HPAA
5-HPAA (0.5 mL) was added to a stirring mixture of aldehyde (1 mmol), malononitrile or ethyl cyanoacetate (1 mmol) and trialkyl phosphite (1 mmol) at room temperature. After stirring for appropriate time (Table 1), water (10 mL) was added to the reaction mixture and washed with EtOAc (3 × 10 mL). The separated organic layer was dried over Na2SO4 and filtered. Evaporation of the filtrate produced almost pure product which was more purified by plate chromatography eluted with n-hexane:EtOAc (1:2). Separated aqueous layer containing the catalyst was evaporated, dried in vacuo at 100 °C for 24 h and reused in the same reaction.
General procedure for the synthesis of 2-amino-4H-chromene substituted with phosphonic acid dialkyl esters in the presence of 5-HPAA
5-HPAA (0.5 mL) was added to a stirred mixture of salicylaldehyde (1.5 mmol), malononitrile or ethyl cyanoacetate (2.5 mmol) and trialkyl phosphite (1 mmol) at room temperature. After stirring for appropriate time (Table 2), water (10 mL) was added to the reaction mixture and the resulting solid (except in 2k)Footnote 1 was filtered. The solid was washed three times with H2O (5 mL). Pure product was obtained from the resulting solid by column chromatography eluted with n-hexane:EtOAc (1:3). The catalyst was recovered after evaporation of the filtrate and drying in vacuo at 100 °C for 24 h.
Spectral data of unknown β-phosphonomalonates
[1-(4-Bromophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1f): (296 mg, 81 %); white solid, mp 102 °C; Rf = 0.44 (n-hexane–EtOAc, 1:1); IR (KBr): 2,242 (CN), 1,000 (P = O) cm−1. 1H NMR (250 MHz, CDCl3): δ 1.16 (t, 3 H, 3JHH = 7.0 Hz), 1.33 (t, 3 H, 3JHH = 7.0 Hz), 3.58 (dd, 1 H, 2JHP = 21.5 Hz, 3JHH = 7.5 Hz), 3.79–4.18 (m, 4 H), 4.56 (t, 1 H, 3JHH = 7.75 Hz), 7.36 (d, 2 H, 3JHH = 8.25 Hz), 7.56 (d, 2 H, 3JHH = 8.0 Hz) ppm; 13C NMR (62.9 MHz, CDCl3): δ 16.1 (d, 3JCP = 4.4 Hz), 16.2 (d, 3JCP = 5.7 Hz), 25.4, 43.9 (d, 1JCP = 144.7 Hz), 63.6 (d, 2JCP = 7.5 Hz), 64.4 (d, 2JCP = 7.5 Hz), 111.0 (d, 3JCP = 11.9 Hz), 111.2 (d, 3JCP = 10.7 Hz), 123.9, 129.4, 131.0, 132.5 ppm; 31P NMR (101 MHz, CDCl3): δ 19.27; MS (EI, 70 eV), m/e: 370 (13 M+), 372 (8 M++2), 233 [8 M+–P(O)(OEt)2], 235 [9 (M++2)–P(O)(OEt)2], 207 (100), 209 (97), 138 (64); Anal. Calcd for C14H16BrN2O3P: C, 45.30; H, 4.34; N, 7.55. Found: C, 44.46; H, 4.15; N, 7.39.
[1-(Pyridin-3-yl)-2,2-dicyanoethyl]phosphonic acid diethyl ester (1i): (265 mg, 91 %); dark orange liquid; Rf = 0.07 (n-hexane–EtOAc, 1:1); IR (KBr): 2,236 (CN), 1,011 (P = O) cm−1; 1H NMR (250 MHz, CDCl3): δ 1.18 (t, 3 H, 3JHH = 6.8 Hz,), 1.33 (t, 3 H, 3JHH = 7.0 Hz), 3.65 (dd, 1 H, 2JHP = 21.6 Hz, 3JHH = 6.8 Hz), 3.92–4.21 (m, 4 H), 4.63 (t, 1 H, 3JHH = 8.5 Hz), 7.39 (t, 1 H, 3JHH = 6.5 Hz), 7.95 (d, 1 H, 3JHH = 6.5 Hz), 8.67 (s, 2 H) ppm; 13C NMR (62.9 MHz, CDCl3): δ 16.1 (d, 3JCP = 5.0 Hz), 16.2 (d, 3JCP = 5.0 Hz), 25.3, 42.1 (d, 1JCP = 144.6 Hz), 63.8 (d, 2JCP = 7.0 Hz), 64.5 (d, 2JCP = 7.0 Hz), 110.8 (d, 3JCP = 10.7 Hz), 111.0 (d, 3JCP = 11.9 Hz), 124.0, 126.7, 136.5, 150.5, 150.8 ppm; 31P NMR (101 MHz, CDCl3): δ 19.03 ppm; MS (EI, 70 eV), m/e (%): 293 (9 M+), 156 [100 M+–P(O)(OEt)2], 138 (94), 130 (25), 111 (93); Anal. Calcd for C13H16N3O3P: C, 53.24; H, 5.50; N, 14.33. Found: C, 52.71; H, 5.32; N, 14.04.
[1,1-Dicyanopentan-2-yl] phosphonic acid diethyl ester (1j): (196 mg, 76 %); yellow liquid; Rf = 0.5 (n-hexane–EtOAc, 1:1); IR (KBr): 2,240 (CN), 1,011 (P = O) cm−1; 1H NMR (250 MHz, CDCl3): δ 1.00 (t, 3 H, 3JHH = 7.0 Hz), 1.37 (t, 6 H, 3JHH = 7.0 Hz), 1.61 (q, 2 H, 3JHH = 7.0 Hz), 1.73–2.10 (m, 2 H), 2.29–2.34 (m, 1 H), 4.16–4.25 (m, 4 H), 4.29–4.35 (m, 1 H) ppm; 13C NMR (62.9 MHz, CDCl3): δ 13.7, 16.3 (d, 3JCP = 5.6 Hz), 20.7 (d, 2JCP = 7.6 Hz), 23.7, 29.2, 37.8 (d, 1JCP = 144.9 Hz), 63.2 (d, 2JCP = 11.0 Hz), 110.7 (d, 3JCP = 5.0 Hz), 112.2 (d, 3JCP = 17.0 Hz) ppm; MS (EI, 70 eV), m/e (%): 259 (5 M++1), 138 (100), 121 [20 M+–P(O)(OEt)2]; Anal. Calcd for C11H19N2O3P: C, 51.16; H, 7.42; N, 10.85. Found: C, 50.99; H, 7.57; N, 10.71.
[1, 1-Dicyanooctan-2-yl] phosphonic acid diethyl ester (1k): (248 mg, 84 %); yellow liquid; Rf = 0.6 (n-hexane–EtOAc, 1:1); IR (KBr): 2,238 (CN), 1,011 (P = O) cm−1; 1H NMR (400 MHz, CDCl3): δ 0.81 (t, 3 H, 3JHH = 6.8 Hz), 1.23–1.25 (m, 6 H), 1.29 (t, 6 H, 3JHH = 7.2 Hz), 1.45–1.50 (m, 2 H), 1.66–1.79 (m, 1 H), 1.86–1.98 (m, 1 H), 2.25–2.34 (m, 1 H), 4.08–4.16 (m, 4 H), 4.36 (dd, 1 H, 3JHP = 13.2 Hz, 3JHH = 3.6 Hz) ppm; 13C NMR (62.9 MHz, CDCl3): δ 13.9, 16.21 (d, 3JCP = 5.0 Hz), 22.3, 23.7, 27.1, 27.2 (d, 3JCP = 6.0 Hz), 28.8, 31.2, 37.7 (d, 1JCP = 143.0 Hz), 63.1 (d, 2JCP = 7.0 Hz), 111.1 (d, 3JCP = 3.0 Hz), 112.3 (d, 3JCP = 17.0 Hz) ppm; MS (EI, 70 eV), m/e (%): 301 (2 M++1), 163 [21 M+–P(O)(OEt)2], 138 (100), 111 (58), 81 (70); Anal. Calcd for C14H25N2O3P: C, 55.99; H, 8.39; N, 9.33. Found: C, 55.36; H, 8.03; N 9.20.
Spectral data of known β-phosphonomalonates
[1-(4-Chlorophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1a): (296 mg, 91 %). yellow solid, mp 98 °C; Rf = 0.53 (n-hexane–EtOAc, 1:1); 1H NMR (250 MHz, CDCl3): δ 1.16 (t, 3 H, 3JHH = 7.0 Hz), 1.33 (t, 3 H, 3JHH = 7.0 Hz), 3.62 (dd, 1 H, 2JHP = 21.5, 3JHH = 7.5 Hz), 3.82–4.19 (m, 4 H), 4.55 (t, 1 H, 3JHH = 7.7 Hz), 7.42 (s, 4 H) ppm.
[1-(4-Chlorophenyl)-2,2-dicyanoethyl] phosphonic acid dimethyl ester (1b): (271 mg, 89 %); white solid, mp 118 °C; Rf = 0.31 (n-hexane–EtOAc, 1:1); 1H NMR (400 MHz, CDCl3): δ 3.61–3.68 (m, 4 H), 3.86 (d, 3 H, 3JHP = 11.2 Hz), 4.51 (t, 1 H, 3JHH = 8.0 Hz), 7.47 (s, 4 H) ppm.
[1-(4-Chlorophenyl)-2,2-dicyanoethyl] phosphonic acid di-iso-propyl ester (1c): (301 mg, 86 %); yellow solid, mp 111 °C; Rf = 0.62 (n-hexane–EtOAc, 1:1); 1H NMR (400 MHz, CDCl3): δ 1.00 (d, 3JHH = 6.4 Hz), 1.32 (d, 3 H, 3JHH = 6.0 Hz), 1.39 (d, 6 H, 3JHH = 6.0 Hz), 3.51 (dd, 1 H, 2JHP = 21.6 Hz, 3JHH = 7.2 Hz), 4.51–4.57 (m, 1 H), 4.75–4.83 (m, 1 H), 7.43–7.50 (m, 4 H) ppm.
[1-(3-Chlorophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1d): (292 mg, 90 %); yellow liquid; Rf = 0.56 (n-hexane–EtOAc, 1:1); 1H NMR (400 MHz, CDCl3): δ 1.20 (t, 3 H, 3JHH = 7.2 Hz), 1.38 (t, 3 H, 3JHH = 7.2 Hz), 3.60 (dd, 1 H, 3JHH = 8.0 Hz, 2JHP = 21.2 Hz), 3.83–3.93 (m, 1 H), 4.03–4.26 (m, 3 H), 4.56 (dd, 1 H, 3JHP = 9.2 Hz, 3JHH = 8.0 Hz), 7.41–7.45 (m, 3 H), 7.50 (s, 1 H) ppm.
[1-(2-Chlorophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1e): (289 mg, 88 %); yellow solid, mp 77 °C; Rf = 0.57 (n-hexane–EtOAc, 1:1); 1H NMR (400 MHz, CDCl3): δ 1.11 (t, 3 H, 3JHH = 7.0 Hz), 1.36 (t, 3 H, 3JHH = 7.0 Hz), 3.75–4.30 (m, 4 H), 4.46 (dd, 1 H, 2JHP = 21.2 Hz, 3JHH = 8.2), 4.61 (t, 1 H, 3JHH = 8.5 Hz), 7.35 (d, 2 H, 3JHH = 4 Hz), 7.47 (s, 1 H), 7.75 (d, 1 H, 3JHH = 5.3 Hz) ppm.
[1-(4-Methoxy)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1g): (257 mg, 81 %); yellow solid, mp 61 °C; Rf = 0.50 (n-hexane–EtOAc, 1:1); 1H NMR (400 MHz, CDCl3): δ 1.17 (t, 3 H, 3JHH = 7.2 Hz), 1.37 (t, 3 H, 3JHH = 7.2 Hz), 3.57 (dd, 1 H, 2JHP = 21.2, 3JHH = 8.0 Hz), 3.84 (s, 3 H), 4.00–4.24 (m, 4 H), 4.51 (dd, 1 H, 3JHP = 8.8 Hz, 3JHH = 8.0 Hz), 6.97 (d, 2 H, 3JHH = 8.8 Hz), 7.41–7.44 (m, 2 H) ppm.
[1-(Furan-2-yl)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1h): (244 mg, 85 %); dark yellow liquid; Rf = 0.42 (n-hexane–EtOAc, 1:1); 1H NMR (250 MHz, CDCl3): δ 1.24–1.37 (m, 6 H), 3.87 (dd, 1 H, 2JHP = 22.7 Hz, 3JHH = 6.5 Hz), 3.98–4.23 (m, 4 H), 4.51 (t, 1 H, 3JHH = 8.7 Hz), 6.44 (s, 1 H), 6.62 (s, 1 H), 7.49 (s, 1 H) ppm.
[1-(4-Chlorophenyl)-2-cyano-2-ethylcarboxylic acid ethyl ester] phosphonic acid diethyl ester (1l): (288 mg, 78 %); light yellow liquid; Rf = 0.46 (n-hexane–EtOAc, 1:1); 1H NMR (400 MHz, CDCl3): δ 1.01–1.09 (m, 2 H), 1.13–1.30 (m, 6 H), 1.35 (t, 1 H, 3JHH = 7.2 Hz), 3.75–3.86 (m, 1 H), 3.90–4.20 (m, 6 H), 4.27 (dd, 1 H, 3JHH = 6.0 Hz, 3JPH = 8.4 Hz), 7.33–7.35 (m, 3 H), 7.46–7.48 (m, 1 H) ppm.
Spectral data of unknown 4-substituted 2-amino-4H-chromenes with phosphonic acid dialkyl esters
Dimethyl 2-amino-3-cyano-4H-chromen-4-yl phosphonate (2b): (249 mg, 89 %); light yellow solid; mp 149 °C; Rf = 0.21 (n-hexane–EtOAc, 1:3); IR (KBr): 3,301 (NH2), 3,114 (NH2), 2,187 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ 3.68 (d, 3 H, 3JHH = 10.4 Hz), 3.80 (d, 3 H, 3JHH = 10.8 Hz), 3.95 (d, 1 H, 2JPH = 18.0 Hz), 4.95 (s, 2 H), 7.02 (d, 1 H, 3JHH = 8.4 Hz), 7.18 (t, 1 H, 3JHH = 7.2 Hz), 7.29–7.31 (m, 1 H), 7.35–7.37 (m, 1 H) ppm; 13C NMR (100 MHz, CDCl3): δ 29.1, 35.0 (d, 1JCP = 148.8 Hz), 50.9, 53.4 (d, 2JCP = 7.2 Hz), 53.8 (d, 2JCP = 7.2 Hz), 116.1, 116.6, 119.3, 125.2, 129.2, 129.5, 149.7, 161.8 ppm; MS (EI, 70 eV), m/e (%): 280 (4 M+–1), 171 [100 M+–P(O)(OEt)2], 42 (88).
Diisopropyl 2-amino-3-cyano-4H-chromen-4-yl phosphonate (2c): (282 mg, 86 %); white solid; mp 158 °C; Rf = 0.05 (n-hexane–EtOAc, 1:3); IR (KBr): 3,355 (NH2), 3270 (NH2), 2,170 (CN) cm−1;1H NMR (400 MHz, DMSO): δ 1.06 (d, 3 H, 3JHH = 6.0 Hz), 1.17 (d, 3 H, 3JHH = 6.4 Hz), 1.18 (d, 3 H, 3JHH = 6.0 Hz), 1.21 (d, 3 H, 3JHH = 6.0 Hz), 3.93 (d, 1 H, 2JPH = 18.0 Hz), 4.33–4.41 (m, 1 H), 4.46–4.54 (m, 1 H), 6.99 (d, 1 H, 3JHH = 8.0 Hz), 7.08–7.15 (m, 3 H), 7.26–7.28 (m, 2 H) ppm; 13C NMR (100 MHz, DMSO): δ 23.8 (d, 3JCP = 5.0 Hz), 24.0 (d, 3JCP = 4.0 Hz), 36.1 (d, 1JCP = 147.9 Hz), 48.4, 71.3 (d, 2JCP = 7.0 Hz), 116.2, 118.4, 118.5, 120.7, 124.5, 129.1, 130.2, 150.4, 162.9 ppm; MS (EI, 70 eV), m/e (%): 336 (3 M+), 171 [100 M+–P(O)(OEt)2].
Diethyl 2-amino-6-methyl-3-cyano-4H-chromen-4-yl phosphonate (2f): (263 mg, 82 %), light yellow solid; mp 171 °C; Rf = 0.03 (n-hexane–EtOAc, 1:3); IR (KBr): 3,340 (NH2), 3,260 (NH2), 3,150 (CN) cm−1; 1H NMR (400 MHz, DMSO): δ 1.13 (t, 3 H, 3JHH = 6.8 Hz), 1.19 (t, 3JHH = 7.2 Hz, 3 H), 2.26 (s, 3 H), 3.86-3.97 (m, 4 H), 4.01 (d, 1 H, 3JPH = 17.6 Hz), 6.90 (d, 3JHH = 8.4 Hz, 1 H), 7.05–7.09 (m, 4 H) ppm; 13C NMR (100 MHz, DMSO): δ 16.6 (d, 3JCP = 5.0 Hz), 20.7, 34.9 (d, 1JCP = 144.9 Hz), 47.8, 62.5 (d, 2JCP = 7.0 Hz), 62.7 (d, 2JCP = 8.0 Hz), 116.1, 117.8, 120.6, 129.6, 130.1, 133.7, 148.3, 148.4, 163.2 ppm; MS (EI, 70 eV), m/e (%): 322 (3 M+), 185 [100 M+–P(O)(OEt)2].
Diethyl 2-amino-8-ethoxy-3-cyano-4H-chromen-4-yl phosphonate (2g): (277 mg, 79 %), white solid, mp 179 °C; Rf = 0.03 (n-hexane–EtOAc, 1:3); IR (KBr): 3,430 (NH2), 3,275(NH2), 2,975 (CN) cm−1; 1H NMR (400 MHz, DMSO): δ 1.11–1.19 (m, 6 H), 1.33 (t, 3JHH = 6.8 Hz, 3 H), 3.87–3.97 (m, 4 H), 4.03 (d, 1 H, 2JPH = 17.6), 4.08–4.11 (m, 2 H), 6.79 (d, 1 H, 3JHH = 7.2 Hz), 6.98 (d, 1 H, 3JHH = 8.4 Hz), 7.04 (t, 1 H, 3JHH = 8.0 Hz), 7.11 (brs, 2H) ppm; 13C NMR (100 MHz, DMSO): δ 15.0, 16.6 (d, 3JCP = 5.0 Hz), 16.7 (d, 3JCP = 4.0 Hz), 35.2 (d, 1JCP = 145.8 Hz), 47.8, 62.6 (d, 2JCP = 7.0 Hz), 62.7 (d, 2JCP = 7.0 Hz), 64.7, 113.5, 119.2, 120.0, 121.2, 140.1, 146.8, 163.1 ppm; MS (EI, 70 eV), m/e (%): 352 (4 M+), 215 [100 M+–P(O)(OEt)2], 187 (23).
2-Amino-4-(diethoxyphosphoryl)-8-ethoxy-4H-chromen-3-carboxylate (2k): (335 mg, 84 %), light yellow oil; Rf = 0.04 (n-hexane–EtOAc, 1:3); IR (KBr): 3,281 (NH2), 3,141 (NH2), 1,689 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.12 (t, 3 H, 3JHH = 7.2 Hz), 1.24 (t, 3 H, 3JHH = 7.2 Hz), 1.31 (t, 3 H, 3JHH = 7.2 Hz), 1.42 (t, 3 H, 3JHH = 7.2 Hz), 3.73–3.83 (m, 1 H), 3.84–3.94 (m, 1 H), 3.98–4.13 (m, 4 H), 4.15–4.28 (m, 2 H), 4.39 (d, 1 H, 2JPH = 19.2 Hz), 6.54 (brs, 2 H), 6.81–6.84 (m, 1 H), 6.91–6.94 (m, 1 H), 7.02 (t, 1 H, 3JHH = 8.0 Hz) ppm;. 13C NMR (100 MHz, CDCl3): δ 14.5, 14.8, 16.3 (d, 3JCP = 6.0 Hz), 16.4 (d, 3JCP = 6.0 Hz), 35.2 (d, 1JCP = 148.9 Hz), 59.6, 62.3 (d, 2JCP = 7.0 Hz), 62.6 (d, 2JCP = 7.0 Hz), 64.7, 112.5, 121.0, 121.1, 124.1, 140.5, 146.7, 161.8, 168.6 ppm; MS (EI, 70 eV), m/e (%): 399 (1 M+), 262 [100 M+–P(O)(OEt)2], 216 (12).
2-Amino-6-chloro-4-(diethoxyphosphoryl)-4H-chromen-3-carboxylate (2l): (334 mg, 86 %); white solid; mp 105 °C; Rf = 0.03 (n-hexane–EtOAc, 1:3); IR (KBr): 3,281 (NH2), 3,141 (NH2), 1,689 (CN) cm−1; 1HNMR (400 MHz, CDCl3): δ 1.15 (t, 3 H, 3JHH = 7.2 Hz), 1.23 (t, 3 H, 3JHH = 7.2 Hz), 1.30 (t, 3 H, 3JHH = 7.2 Hz), 3.83–4.08 (m, 4 H), 4.11–4.26 (m, 2 H), 4.33 (d, 1 H, 2JPH = 20.0 Hz), 6.61 (brs, D2O exchangeable, 2 H), 6.87–6.91 (m, 1 H), 7.14–7.18 (m, 1 H), 7.29–7.31 (m, 1 H) ppm; 13CNMR (100 MHz, CDCl3): δ 14.5, 16.3, 16.4, 35.0 (d, 1JCP = 146.9 Hz), 59.7, 62.5 (d, 2JCP = 7.0 Hz), 62.8 (d, 2JCP = 8.0 Hz), 117.2, 121.7, 121.8, 128.2, 129.1, 129.2, 149.3, 161.7, 168.3 ppm; MS (EI, 70 eV), m/e (%): 389 (1 M+), 252 [100 M+-P(O)(OEt)2], 206 (43).
Spectral data of known 4-substituted 2-amino-4H-chromenes with phosphonic acid dialkyl esters
Diethyl 2-amino-3-cyano-4H-chromen-4-yl phosphonate (2a): (280 mg, 91 %); white solid, mp 140–142 °C; Rf = 0.06 (n-hexane–EtOAc, 1:3); 1HNMR (400 MHz, CDCl3): δ 1.19 (t, 3 H, 3JHH = 7.0 Hz), 1.33 (t, 3 H, 3JHH = 7.0 Hz), 3.88 (d, 1 H, 2JPH = 18.0 Hz), 3.94–4.17 (m, 4 H), 5.07 (s, 2 H), 6.96 (d, 1 H, 3JHH = 8.3 Hz), 7.13 (t, 1 H, 3JHH = 7.3 Hz), 7.22–7.34 (m, 2 H) ppm.
Diethyl 2-amino-8-methoxy-3-cyano-4H-chromen-4-yl phosphonate (2e): (297 mg, 90 %); white solid, mp 170 oC; Rf = 0.04 (n-hexane–EtOAc, 1:3); 1H NMR (400 MHz, CDCl3): δ 1.21 (t, 3 H, 3JHH = 6.8 Hz), 1.37 (t, 3 H, 3JHH = 7.2 Hz), 3.88 (s, 3 H), 3.91 (d, 1JPH = 16.0 Hz, 1 H), 3.95–4.04 (m, 2 H), 4.12–4.20 (m, 2 H), 4.93 (s, 2 H), 6.88 (d, 1 H, 3JHH = 5.2 Hz), 6.95 (d, 1 H, 3JHH = 7.6 Hz), 7.10 (t, 1 H, 3JHH = 8 Hz) ppm.
Results and discussion
Initially, to optimize the quantity of ionic liquid required, the coupling reaction between 4-chlorobenzaldehyde, malononitrile and triethyl phosphite was carried out in the presence of 0.1, 0.25 and 0.5 mL of 5-HPAA at room temperature. In these cases, the desired β-phosphonomalonate was formed in 65, 76 and 90 % isolated yields after 20, 15 and 15 min, respectively. According to these results, to establish the generality of this ionic liquid for the synthesis of a series of β-phosphonomalonates, the coupling reaction of various structurally diverse aldehydes with malononitrile and trialkyl phosphites was investigated using 0.5 mL of 5-HPAA as the optimal amount (Scheme 1; Table 1).

Synthesis of β-phosphonomalonates in the presence of 5-HPAA
As is obvious from Table 1, the catalytic one-pot reaction of 4-chlorobenzaldehyde and malononitrile proceeded well with trialkyl phosphites such as triethyl/trimethyl/tri-iso-propyl phosphites (entries 1–3). These results demonstrate that both the yields and the reaction times are relatively independent of the phosphorus compounds. Differently substituted benzaldehydes with electron-donating and electron-withdrawing groups underwent successful Knoevenagel-phospha-Michael reaction with malononitrile and triethyl phosphite (entries 4–7). The catalyst was compatible with functional groups such as Cl, Br and O–Me. No competitive nucleophilic methyl ether cleavage was observed in the substrate possessing an aryl-O–Me group (entry 7), despite the strong nucleophilicity of phosphites. Acid-sensitive aldehydes, such as furan-2-carbaldehyde and pyridine-3-carbaldehyde underwent smooth reactions without any decomposition or polymerization under the present reaction conditions (entries 8 and 9). This method is also applicable for the synthesis of β-phosphonomalonates from the reaction of triethyl phosphite with aliphatic aldehydes and malononitrile (entries 10 and 11). In addition to malononitrile, the reaction of ethyl cyanoacetate and 4-chlorobenzaldehyde as other in situ generated Michael acceptors with triethyl phosphite was also examined (entry 12). The results showed that the reaction worked well and the desired product was obtained in 78 % yield.
It should be noted that no competitive side reactions, such as the formation of α-hydroxyphosphonates, were observed in these transformations. These observations prompted us to study the generation of α-hydroxyphosphonate as a probable intermediate in these reactions. We have found that the treatment of 4-chlorobenzaldehyde with triethyl phosphite in the presence of 5-HPAA at room temperature did not produce any significant amount of the corresponding α-hydroxyphosphonate even after 24 h (IR, NMR, MS).
In order to study the possibility of the formation of α,β-unsaturated malonate as another intermediate in the initial Knoevenagel reaction, the reaction of 4-chlorobenzaldehyde and malononitrile in the presence of 5-HPAA at room temperature was also investigated. The results showed that the desired α,β-unsaturated malonate was isolated in 95 % yield after 5 min.
These observations delineate the tandem Knoevenagel-phospha-Michael reaction for the synthesis of β-phosphonomalonates in 5-HPAA. Thus, Knoevenagel reaction between the aldehydes and malononitrile produces an initial α,β-unsaturated malonate. Subsequent phospha-Michael addition of trialkyl phosphites leads to the formation of the desired products. It is noteworthy to mention that reports on the synthesis of β-phosphonomalonates are mainly focused on the two-pot procedures and the report for the one-pot synthesis of these compounds directly from simple and readily available starting materials is rare in the literature [22, 46].
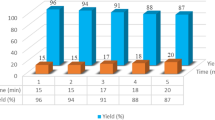
The reusability of the catalyst was also tested in the synthesis of [1-(4-chlorophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1a) from the reaction of 4-chlorobenzaldehyde, malononitrile and triethyl phosphite. After each run, EtOAc was added to the reaction mixture and washed with water. 5-HPAA was obtained by evaporation of the solvent of aqueous layer. After drying in vacuo at 100 °C, 5-HPAA was reused for a consecutive run under the same reaction conditions. It was seen that 5-HPAA displayed good reusability after three runs (Fig. 1).

Reusability of 5-HPAA for the synthesis of [1-(4-chlorophenyl)-2,2-dicyanoethyl] phosphonic acid diethyl ester (1a)
Successful application of 5-HPAA as a reusable ionic liquid for the one-pot synthesis of β-phosphonomalonates via P–C bond formation, encouraged us to study the applicability of this method for the coupling reaction of salicylaldehyde, malononitrile or ethyl cyanoacetate and trialkyl phosphites for the synthesis of 4-substituted 2-amino-4H-chromenes with phosphonic acid dialkyl esters (Scheme 2; Table 2).

Synthesis of 4-substituted 2-amino-4H-chromenes in the presence of 5-HPAA
4-Substituted 2-amino-4H-chromenes are introduced as analogues of tumor antagonist HA 14-1 (Scheme 3) [61, 62]. HA 14-1 is a new class of small molecules that exhibit a binding activity for the surface pocket of the cancer-implicated Bcl-2 protein and induce apoptosis or programmed cell death in follicular lymphoma B cells and leukemia HL-60 cells [61, 62]. A survey of the literature indicates that there is no report aimed at the synthesis of 4-substituted 2-amino-4H-chromenes with phosphonic acid dialkyl esters utilizing ionic liquids.

HA 14-1
As shown in Table 2, salicylaldehyde underwent coupling reaction with malononitrile and triethyl/trimethyl/tri-iso-propyl phosphite and produced the desired products in 86–91 % yields in the presence of 5-HPAA at room temperature (entries 1–3). Coupling reaction of substituted salicylaldehydes bearing electron-releasing and electron-withdrawing groups with malononitrile or ethyl cyanoacetate and triethyl phosphite proceeded well and gave the corresponding products in 78–90 % yields under the same reaction conditions (entries 4–12).
We have also suggested a plausible mechanism for the one-pot reaction of aldehydes, malononitrile and trialkyl phosphites in the presence of 5-HPAA (Scheme 4).

Suggested mechanism for the synthesis of β-phosphonomalonates in the presence of 5-HPAA
The process represents a typical tandem reaction, in which aldehydes first condensed with malononitrile to form the Knoevegel product followed by Michael addition with trialkyl phosphite to produce β-phosphonomalonates. In this reaction, two spontaneous interactions between NH3 + and OH of 5-HPAA with carbonyl groups of aldehydes and hydrogen atoms of the active methylene group of malononitrile took place. These interactions activated the reactants and facilitated the formation of the product. In the case of salicylaldehyde, cyclization reaction occurred in the Knoevegel product to form imino chromene 3 (Scheme 5).

Imino chromene
Compound 3 underwent Michael addition with trialkyl phosphite as a phosphorus nucleophile to produce 4-substituted 2-amino-4H-chromenes with phosphonic acid dialkyl esters (2).
In order to examine the effect of 5-HPAA as an ionic liquid for the synthesis of β-phosphonomalonates, the coupling reaction between 4-chlorobenzaldehyde, malononitrile and triethyl phosphite in the absence of ionic liquid at room temperature was considered. This reaction led to the formation of the desired product (1a) in low yield (40 %) after 20 h due to the formation of a mixture of by-products (Table 3, entry 1). A similar reaction in the presence of [CH3(CH2)4NH3 +][−OAc] proceeded with a longer reaction time (4 h) compared with 5-HPAA (15 min) to produce the desired product in 85 % yield (Table 1, entries 2 and 3). These results showed the specific role of OH group of 5-HPAA in imparting the catalytic property and indicate that 5-HPAA can act as a functionalized or task-specific ionic liquid in this method. We have also found that shortening of the alkyl chain in [HO(CH2)nNH3 +][−OAc] has a small influence on the efficiency of ionic liquid for the synthesis of 1a (Table 3, entries 4 and 5). More studies showed that when ethyl cyanoacetate was used instead of malononitrile, the reaction rate and yield of the product (1l) decreased with shortening the length of the alkyl chain (Table 3, entries 6 and 7).
Conclusions
In conclusion, we have successfully developed a simple one-pot protocol to generate P–C bonds via a tandem Knoevenagel-phospha-Michael reaction in the presence of an ionic liquid for the first time. This simple procedure allows a series of β-phosphonomalonates to be synthesized from the reaction of aryl/heteroaryl/alkyl aldehydes, malononitrile and trialkyl phosphites at room temperature. This method is also applicable for the synthesis of 4-substituted 2-amino-4H-chromenes with phosphonic acid dialkyl esters from the coupling reaction of salicylaldehyde, malononitrile/ethyl cyanoacetate and trialkyl phosphites. Short reaction times, ease of preparation of 5-HPAA as a cost-effective and task-specific ionic liquid and its reusability as an environmentally benign promoter are the other advantageous of the present protocol.
Notes
After adding H2O (10 mL) to the reaction mixture, it was washed with EtOAc (3 × 10 mL). The separated organic layer was dried over Na2SO4 and filtered. Evaporation of the filtrate produced almost pure product which was purified by plate chromatography eluted with n-hexane:EtOAc (1:3).
References
M.C. Allen, W. Fuhrer, B. Tuck, R. Wade, J.M. Wood, J. Med. Chem. 32, 1652 (1989)
D.V. Patel, K. Rielly-Gauvin, D.E. Ryono, Tetrahedron Lett. 31, 5587 (1990)
B. Stowasser, K.H. Budt, J.Q. Li, A. Peyman, D. Ruppert, Tetrahedron Lett. 33, 6625 (1992)
P. Kafarski, B. Lejczak, Phosphorus. Sulfur Silicon Relat. Elem. 63, 193 (1991)
E.K. Baylis, C.D. Campbell, J.G. Dingwall, J. Chem. Soc. Perkin Trans. I 2845 (1984)
F.R. Atherton, C.H. Hassal, R.W. Lambert, J. Med. Chem. 29, 29 (1986)
B.E. Maryanoff, A.B. Reitz, Chem. Rev. 89, 863 (1989)
N.L. Shipkowitz, R.R. Bower, R.N. Appel, C.W. Nordeen, L.R. Overby, W.R. Roderick, J.B. Schleicher, A.M. Von Esch, Appl. Microbiol. 26, 264 (1973)
J.C.H. Mao, E.E. Robishaw, Biochemistry 26, 264 (1973)
C.L.K. Sabourin, J.M. Reno, J.A. Boezi, Arch. Biochem. Biophys. 187, 96 (1978)
J.C.H. Mao, E.R. Otis, A.M. Von Esch, T.R. Herrin, Antimicrob. Agents Chemother. 27, 197 (1985)
E. Sandstrom, J.C. Kaplan, R.E. Byington, M.S. Hirsch, Lancet 1, 1480 (1985)
V.D. Patel, R.J. Schmidt, S.A. Biller, E.M. Gordon, S.S. Robinson, V. Manne, J. Med. Chem. 38, 2906 (1995)
K. Green, Tetrahedron Lett. 30, 4807 (1989)
R.R. Hindersinn, R.S. Ludington, J. Org. Chem. 30, 4020 (1965)
Z. Jiang, Y. Zhang, W. Ye, C.-H. Tan, Tetrahedron Lett. 48, 51 (2007)
A.N. Pudovik, I.V. Konovalova, Synthesis 2, 81 (1979)
D. Enders, A. Saint-Dizier, M.I. Lannou, A. Lenzen, Eur. J. Org. Chem. 29 (2006)
R.C. Miller, J.S. Bradley, L.A. Hamilton, J. Am. Chem. Soc. 78, 5299 (1956)
R. Bodalski, K. Pietrusiewicz, Tetrahedron Lett. 13, 4209 (1972)
D. Simoni, F.P. Invidiata, M. Manferdini, I. Lampronti, R. Rondanin, M. Roberti, G.P. Pollini, Tetrahedron Lett. 39, 7615 (1998)
M. Hosseini-Sarvari, S. Etemad, Tetrahedron 64, 5519 (2008)
M.O. Shulyupin, M.A. Kazankova, I.P. Beletskaya, Org. Lett. 4, 761 (2002)
Q. Xu, L.-B. Han, Org. Lett. 8, 2099 (2006)
D. Semenzin, G. Etemad-Moghadam, D. Albouy, O. Diallo, M. Koenig, J. Org. Chem. 62, 2414 (1997)
L.-B. Han, C.-Q. Zhao, J. Org. Chem. 70, 10121 (2005)
J.R.A. Stockland, R.I. Taylor, L.E. Thompson, P.B. Patel, Org. Lett. 7, 851 (2005)
M.R. Mahran, W.M. Abdou, Heteroat. Chem. 3, 93 (1992)
T. Welton, Chem. Rev. 99, 2071 (1999)
P. Wasserscheid, W. Keim, Angew. Chem. Int. Ed. 39, 3772 (2000)
T. Welton Coord, Chem. Rev. 248, 2459 (2004)
X. Mi, S. Luo, J.P. Cheng, J. Org. Chem. 70, 2338 (2005)
R.V. Hangarge, D.V. Jarikoteb, M.S. Shingare, Green Chem. 4, 266 (2002)
V. Singh, S. Kaur, V. Sapehiyi, Catal. Commun. 6, 57 (2005)
S. Sobhani, M. Faal Maleki, Synlett. 383 (2010)
S. Sobhani, Z. Tashrifi, Tetrahedron 66, 1429 (2010)
S. Sobhani, Z. Tashrifi, Heteroat. Chem. 20, 109 (2009)
S. Sobhani, Z. Tashrifi, Synth. Commun. 39, 120 (2009)
S. Sobhani, A. Vafaee, Tetrahedron 65, 7691 (2009)
S. Sobhani, E. Safaei, M. Asadi, F. Jalili, J. Organomet. Chem. 693, 3313 (2008)
S. Sobhani, E. Safaei, M. Asadi, F. Jalili, Z. Tashrifi, J. Porphyrins Phthalocyanines 12, 849 (2008)
S. Sobhani, A. Vafaee, Synthesis 1909 (2009)
S. Sobhani, A. Vafaee, J. Iran. Chem. Soc. 7, 227 (2010)
S. Sobhani, S. Rezazadeh, Synlett 1485 (2010)
S. Sobhani, S. Rezazadeh, J. Iran. Chem. Soc. 8, 198 (2011)
S. Sobhani, Z. Pakdin Parizi, Tetrahedron 67, 3540 (2011)
S.J. Zhang, X.L. Yuan, Y.H. Chen, Y.Q. Zhang, Chin Patent 10069408.5 (2005)
N. Bicak, J. Mol. Liq. 116, 15 (2005)
C. Yue, A. Mao, Y. Wei, M. LÜ, Catal. Commun. 9, 1571 (2008)
A. Alizadeh, M.M. Khodaei, A. Eshghi, J. Org. Chem. 75, 8295 (2010)
H.R. Shaterian, M. Arman, F. Rigi, J. Mol. Liq. 158, 145 (2011)
K.A. Kurnia, F. Harris, C.D. Wilfred, M.I. Abdul Mutalib, T. Murugesan, J. Chem. Thermodyn. 41, 1069 (2009)
X. Yuan, S. Zhang, J. Liu, X. Lu, Fluid Phase Equilib. 257, 195 (2007)
X. Yuan, S. Zhang, X. Lu, J. Chem. Eng. Data 52, 596 (2007)
G. Lilia Ben, Z. Hedi, Phosphorus, Sulfur Silicon Relat. Elem. 157, 153 (2000)
W.M. Abdou, M.D. Khidre, M.R. Mahran, J. Prakt. Chem. 332, 1029 (1990)
A.A. Fahmy, N.A. Ismail, T.S. Hafez, Phosphorus, Sulfur Silicon Relat. Elem. 66, 201 (1992)
T.G. Rymareva, V.B. Sandakov, B.A. Khaskin, V.K. Promonenkov, T.I. Koroleva, Zh.Obshch.Khim. 52, 220 (1982)
P. Jayashree, G. Shanthi, P.T. Perumal, Synlett 6, 917 (2009)
S. Narayana Murthy, B. Madhav, V. Prakash Reddy, Y.V.D. Nageswar, Tetrahedron Lett. 51, 3649 (2010)
J. Skommer, D. Wlodkowic, M. Matto, M. Eray, J. Pelkonen, Leukemia Res. 30, 322 (2006)
J.L. Wang, D. Liu, Z. Zhang, S. Shan, X. Han, S.M. Srinvasula, C.M. Croce, E.S. Alnemeri, Z. Huang, Proc. Natl. Acad. Sci. USA 97, 7124 (2000)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sobhani, S., Honarmand, M. 5-Hydroxypentylammonium acetate as a reusable ionic liquid catalyzes tandem Knoevenagel-phospha-Michael reaction of aldehydes, malononitrile and phosphites. J IRAN CHEM SOC 9, 661–669 (2012). https://doi.org/10.1007/s13738-012-0088-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-012-0088-1