
Abstract
Rhabdomyolysis is characterized by the acute breakdown of skeletal muscle, resulting in the release of muscle cell contents, subsequent myoglobinuria, and in severe cases, acute renal failure. A number of etiologies have been identified in acute rhabdomyolysis, in which drugs and trauma account for the majority of cases. One etiological category that is commonly overlooked in the adult population is an underlying genetic defect. This may be challenging to diagnose due to its rarity in the adult demographic and the marked heterogeneity, often requiring a high level of clinical suspicion before investigation is pursued. Once diagnosed, however, appropriate steps can be taken to reduce future episodes of rhabdomyolysis, further renal injury, and other systemic complications. Here, we report a case of an adult patient presenting with acute rhabdomyolysis secondary to McArdle disease, a genetic disease causing defective glycogenolysis. The case highlights the importance of recognizing the potential of undiagnosed “pediatric” disorders in adulthood and particularly for underlying genetic causes of rhabdomyolysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The underlying etiology of acute rhabdomyolysis is broad. Drugs and trauma account for up to 80% of cases in the adult population [1, 2]. For the small percentage of rhabdomyolysis due to genetic causes, most are diagnosed by adolescence [3]. In this case report, we present an adult patient with rhabdomyolysis secondary to McArdle disease, a genetic and neuromuscular disorder well recognized in the pediatric population. The case highlights the importance of gathering a careful history of symptoms related to physical activity and recognizing the potential of undiagnosed “pediatric” disorders presenting in adulthood.
Case report
A 43-year-old man presented with generalized myalgias. This started 1 week prior to admission while vacationing in Mexico, where he suffered a near drowning incident. A few hours later, he began to experience diffuse muscle soreness and several episodes of “tea” colored urine. He was seen by a physician there, who told him that his “kidneys were failing.” The patient returned to our institution 8 days after symptom onset. Past medical history is significant for hypertension and diabetes mellitus type II. There is no family history of similar symptoms. His only home medication was metformin 500 mg twice daily. With further questioning, the patient endorsed daily synthetic marijuana use while on vacation.
Vitals were significant for known hypertension but otherwise normal. Neurological examination was normal. Laboratory studies were notable for the following: sodium 133 mmol/L, potassium 6.5 mmol/L, chloride 99 mmol/L, bicarbonate 7 mmol/L, BUN 219 mg/dL, creatinine 25.27 mg/dL, glucose 103 mg/dL, phosphorus 17.1 mg/dL, uric acid 15.1 mg/dL, and creatinine kinase 4569 U/L. Outside records revealed normal baseline BUN and creatinine 5 months prior, and sparse records from Mexico revealed a BUN of 342 mg/dL and creatinine of 32.3 mg/dL 8 days prior. Urinalysis was as follows: specific gravity 1.011, pH 5.5, large blood, trace protein, and 0–3 red blood cells. Urine drug screen was negative. A CT of the chest, abdomen, and pelvis was negative for abnormalities including malignancy. After emergent temporary hemodialysis and aggressive intravenous volume repletion, the patient showed signs of renal recovery with eventual normalization of chemistries.
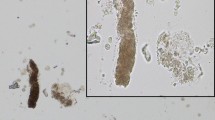
With further probing, the patient retrospectively recalled a pattern of tea colored urine with preceding exercise. He learned the limits of his physical activity from a young age and was able to avoid such episodes throughout his life. His urine myoglobin later returned at 972 μg/L (reference range <28 μg/L). A muscle biopsy was pursued and demonstrated a complete absence of the myophosphorylase enzyme activity (Fig. 1). Genetic testing confirmed a homozygous mutation of the PYGM gene.

Left figure adapted from Dubowitz et al. [26]
Left normal fiber pattern in control sample. Right muscle biopsy of the left quadriceps from our patient stained for phosphorylase showing a total absence of activity.
Discussion
Metabolic myopathies comprise of a heterogenous group of disorders, which are split into three broad categories: glycogen storage diseases, fatty acid metabolism defects, and mitochondrial disorders. Together, they span a variety of clinical presentations, from infantile-onset multi-organ involvement to adult-onset isolated myopathies. Still, they remain a diagnostic challenge, because common to this diverse collection of diseases is their shared syndromic complaint of exercise intolerance and recurrent myoglobinuria. Nonetheless, there are distinct clinical patterns unique to each disorder, which becomes essential to recognize as they can guide diagnostic testing.
Our patient was diagnosed with myophosphorylase deficiency, also known as McArdle disease, or Glycogen Storage Disease (GSD) Type V. This is the most common disorder of glycogen metabolism caused by a homozygous mutation in the PYGM gene, leading to a partial or complete absence of the myophosphorylase enzyme [4]. The enzyme catalyzes the breakdown of muscle glycogen by removing 1,4 glycosyl units from the glycogen molecule, which liberates glucose-1-phosphate (Fig. 2). Its conversion to glucose-6-phosphate then becomes a participant of glycolysis. The enzyme deficit leads to a blockage of glycolysis and subsequent muscle necrosis, resulting in generalized myalgias, myoglobinuria, and, in some cases, rhabdomyolysis with acute kidney injury (AKI). Muscle complaints usually present from a young age, sometimes mislabeled as “growing pains,” which can delay diagnosis until adulthood. Symptoms are induced within minutes following isometric muscle activity. This is in contrast with symptoms of lipid metabolism disorders, which appear after hours of prolonged exercise. The pathognomonic feature of McArdle disease is the so-called “second wind” phenomenon [5]. This denotes a sudden improvement in exercise capacity after ingesting a glucose load prior to exercise. Conversely, the “out-of-wind” phenomenon occurs when glucose intake preceding exercise exacerbates muscle symptoms, indicative of another glycogen storage disorder caused by the absence of glycolytic enzyme AB phosphofructokinase (GSD Type VII). The two opposing mechanisms are determined by the location of the enzyme deficit, with the former representing enzyme blockage upstream of glucose catabolism and the latter with blockage downstream of the entry of glucose into glycolysis [6]. Thus, obtaining a history of a “second wind” is essential in any patient presenting with exercise intolerance.

Adapted from Lucia et al. [6]
Schematic representation of skeletal muscle glycogen metabolism. The figure denotes the steps that are affected in various glycogen storage diseases. GSD glycogen storage disease, UDP uridine diphosphate.
Disorders of fatty acid oxidation often present in infancy with multi-organ involvement, but milder forms of the condition present in adolescence with predominantly myopathic complaints. Carnitine palmitoyltransferase II (CPT II) deficiency is the most common lipid metabolism disorder, and symptoms primarily affect skeletal muscle, presenting as myalgias associated with myoglobinuria. However, these symptoms are not only provoked by prolonged exercise, but also infections, fasting, emotional stress, drugs, alcohol, and cold exposure or heat exposure [7]. The CK level is usually normal outside of episodes of muscle injury, compared to the elevated baseline CK level in McArdle disease (mean 2000–3000 U/L). Since a muscle biopsy in CPT II deficiency is often normal, a fasting blood acylcarnitine profile is the first-line investigation. The diagnosis is confirmed by a combination of enzyme assays and genetic testing [8]. Sometimes, symptoms and muscle biopsies are inadequate in discriminating between the different lipid metabolism disorders. Very long-chain acyl-coA deficiency (VLCAD), for example, has a similar presentation to CPT II deficiency. In both diseases, symptoms are induced by the same provocation factors. The diagnosis of VLCAD is made by blood acyl carnitines by tandem mass spectrometry, showing an abnormal long-chain acyl carnitine with tetradecenoylcarnitine (C14:1) as the predominant species [9].
In comparison to the previous categories, mitochondrial disorders classically occur within a constellation of symptoms. Mitochondrial diseases may be inherited maternally or via autosomal dominant, autosomal recessive, or X-linked fashions. They are clinically heterogeneous, can affect any organ system at any age, and are typically progressive. Symptoms often involve the central nervous and cardiovascular systems. Kearns–Sayre syndrome, for example, is defined by the classical triad of age of onset before 20, extraocular muscle weakness, and pigmentary retinopathy, plus one of the following: ataxia, protein level greater than 100 mg/dL in cerebrospinal fluid, or cardiac conduction abnormalities [10]. Mitochondrial disease can be investigated either molecularly by gene panel or whole-exome sequencing, or by pathologic findings on muscle biopsy of abnormal respiratory chain function or the presence of ragged red fibers. However, a normal biopsy may not definitely rule out a mitochondrial disorder. A variety of metabolites on urine or plasma amino acids can suggest mitochondrial dysfunction, and generalized aminoaciduria can support mitochondrial tubular dysfunction [11].
Other possibilities were considered for our patient prior to the muscle biopsy and genetic confirmation. His CK level on admission to our center was not significantly elevated to the degree which one would expect with rhabdomyolysis, as the frequency of AKI in rhabdomyolysis is usually low when CK values are less than 15,000–20,000 U/L [12]. It is likely that his rhabdomyolysis was already improving along with his AKI based on the records obtained from Mexico. CK levels typically peak at 24–36 h after the inciting event and then decreases back to baseline at a rate of 40% per day [13]. Presuming that the patient was already recovering from the drowning incident 8 days prior, his presenting CK level would have been consistent with the rate of downward trend. Nonetheless, the prospect of drug-induced AKI and rhabdomyolysis was entertained given his metformin and synthetic marijuana use. However, evidence for either drug as a cause for acute rhabdomyolysis is only limited to case reports. One case described the association of metformin to rhabdomyolysis in the context of a combined overdose of metformin, ramipril, and alcohol [14]. Another described its association during the concomitant use with an antipsychotic drug [15]. To our knowledge, there have been no reports in the literature describing a direct link between metformin and rhabdomyolysis, although there have been case reports connecting other antidiabetic agents to rhabdomyolysis, such as sitagliptin, rosiglitazone, pioglitazone, and troglitazone [16,17,18,19]. In all of the cases, the described association has occurred in patients who were also taking other medications in addition to the antidiabetic agent of interest, including ones notoriously known to cause rhabdomyolysis, such as statins and fibrates. This observation led some authors to speculate that the high doses of antidiabetic medications had contributed to a decreased renal function, thereby precipitating rhabdomyolysis by increasing circulating levels of the statin or fibrate. In one study involving over 125,000 patients, those who had used antidiabetic medications did not experience an increased risk of a myopathic event compared to those who had not [20]. The overarching theme appears to be that rhabdomyolysis is more likely to occur as a consequence of drug–drug interactions and toxicities that accumulate as a result, rather than direct insult by an antidiabetic agent alone.
Synthetic cannabinoids are a blend of herbal and chemical compounds that imitate the effects of marijuana. They are becoming more popular due to the high potency that is achieved when smoked but also to their difficulty in being detected in a urine drug screen [21]. There have been several reports connecting synthetic marijuana to AKI in the form of acute tubular necrosis [22] and/or rhabdomyolysis [23,24,25]. It is also possible that the synthetic cannabinoids acted as a “second hit” for his genetic disease to manifest clinically. A renal biopsy was considered given the several possibilities mentioned above. This was not pursued, because differentiating rhabdomyolysis from acute tubular necrosis was not clinically necessary as it would have not have changed management. Moreover, his kidney function was already showing improvement by the time that a renal biopsy was considered. Ultimately, his presentation and clinical history led us to obtain the muscle biopsy and confirm the underlying diagnosis of McArdle disease.
In conclusion, this case serves as a reminder of the potential for undiagnosed “pediatric” diseases in adulthood. The careful collection of an exercise history was a clinical key for an eventual diagnosis of an inherited myopathy. Although treatment for McArdle disease at this time is supportive, patient education about diet, exercise, and avoiding drugs of abuse is important. Adequate hydration, carbohydrate intake before exercise, appropriate exercise instruction that includes gradual warm-up, use of the second wind phenomenon, and avoidance of certain types of activity such as sprinting and isometric exercise can help to prevent future episodes of rhabdomyolysis and kidney injury.
References
Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine. 1982;61(3):141–52.
Ward MM. Factors predictive of acute renal failure in rhabdomyolysis. Arch Intern Med. 1988;148(7):1553–7.
Elsayed EF, Reilly RF. Rhabdomyolysis: a review, with emphasis on the pediatric population. Pediatr Nephrol. 2010;25(1):7–18.
Rubio JC, Lucia A, Fernández-Cadenas I, et al. Novel mutation in the PYGM gene resulting in McArdle disease. Arch Neurol. 2006;63(12):1782–4.
Pearson CM, Rimer DG, Mommaerts WFHM. A metabolic myopathy due to absence of muscle phosphorylase. Am J Med. 1961;30(4):502–17.
Lucia A, Nogales-Gadea G, Pérez M, et al. McArdle disease: what do neurologists need to know? Nat Clin Pract Neurol. 2008;4(10):568–77.
Quinlivan R, Jungbluth H. Myopathic causes of exercise intolerance with rhabdomyolysis. Dev Med Child Neurol. 2012;54(10):886–91.
Thuillier L, Rostane H, Droin V, et al. Correlation between genotype, metabolic data, and clinical presentation in carnitine palmitoyltransferase 2 (CPT2) deficiency. Hum Mutat. 2003;21(5):493–501.
Laforêt P, Vianey-Saban C. Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscul Disord. 2010;20(11):693–700.
Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med. 2013;45(1):4–16.
Scalco RS, Gardiner AR, Pitceathly RDS. Rhabdomyolysis: a genetic perspective. Orphanet J Rare Dis. 2015;10(1):51.
Bosch X, Poch E, Grau JM, et al. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361(1):62–72.
Scalco RS, Snoeck M, Quinlivan R, et al. Exertional rhabdomyolysis: physiological response or manifestation of an underlying myopathy? BMJ Open Sport Exerc Med. 2016;2:e000151.
Galea M, Jelacin N, Bramham K, et al. Severe lactic acidosis and rhabdomyolysis following metformin and ramipril overdose. Br J Anaesth. 2007;98(2):213–5.
Ursini F, Succurro E, Grembiale A, et al. Acute rhabdomyolysis during treatment with amisulpride and metformin. Eur J Clin Pharmacol. 2010;66(3):321–2.
Kao DP, Kohrt HE, Kugler K. Renal failure and rhabdomyolysis associated with sitagliptin and simvastatin use. Diabet Med. 2008;25(10):1229–30.
Yokayama M, Izumiya Y, Yoshizawa M, et al. Acute rhabdomyolysis associated with troglitazone. Diabetes Care. 2000;23:421–2.
Ledl M, Hohenecker J, Francesconi C. Acute myopathy in a type 2 diabetic patient on combination therapy with metformin, fenofibrate, and rosiglitazone. Diabetologia. 2005;48:1996–8.
Slim R, Salem CB, Zamy M, et al. Pioglitazone-induced acute rhabdomyolysis. Diabetes Care. 2009;7:e84.
Koro CE, Sowell MO, Stender M, et al. The risk of myopathy associated with thiazolidinediones and statins in patients with type 2 diabetes: a nested case control analysis. Clin Ther. 2008;30(3):535–42.
Harris CR, Brown A. Synthetic cannabinoid intoxication: a case series and review. J Emerg Med. 2013;44(2):360–6.
Bhanushali GK, Jain G, Fatima H, et al. AKI associated with synthetic cannabinoids: a case series. Clin J Am Soc Nephrol. 2012;8(4):523–6.
Durand D, Delgado LL, de la Parra-Pellot DM, et al. Psychosis and severe rhabdomyolysis associated with synthetic cannabinoid use: a case report. Clin Schizophr Relat Psychoses. 2013;8(4):205–8.
Paul ABM, Simms L, Paul AE, et al. Not safe for consumption: synthetic cannabinoids causing fatal acute rhabdomyolysis in two young men. Int J Case Rep Images. 2016;7:431–5.
Zhao A, Tan M, Maung A, et al. Rhabdomyolysis and acute kidney injury requiring dialysis as a result of concomitant use of atypical neuroleptics and synthetic cannabinoids. Case Rep Nephrol. 2015;2015:235982.
Dubowitz V, et al. Muscle biopsy: a practical approach. Amsterdam: Elsevier Health Sciences; 2013. p. 89.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Human and animal rights statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
About this article

Cite this article
Zhao, X., Li, A., Soni, M. et al. McArdle disease: a “pediatric” disorder presenting in an adult with acute kidney injury. CEN Case Rep 6, 156–160 (2017). https://doi.org/10.1007/s13730-017-0265-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-017-0265-2