
Abstract
Rhabdomyolysis is a common cause of acute kidney injury (AKI) that is usually triggered by trauma. However, less common causes of rhabdomyolysis may precipitate AKI as well, possibly representing a diagnostic challenge even for the experienced nephrologist. Genetic defects of muscle metabolism represent one of these causes and can be overlooked in adults, since these diseases usually become apparent in childhood. We present here a case in which an adult patient with severe exertional rhabdomyolysis leading to AKI was finally diagnosed with a genetic defect of lipid metabolism. A 41-year-old patient was brought to our attention because of AKI and pigmenturia after strenuous physical effort. At admission, the patient was over-hydrated with a weight increase of 3 kg in few days. Laboratory examination showed creatinine of 8.7 mg/dl, along with increased myoglobin and CPK. Urinalysis was positive for haemoglobin and proteins, while urinary sediment analysis did not demonstrate any red blood cell but rather “muddy-brown” casts and tubular cells. Urine output was forced and the patient completely recovered renal function. Genetic analysis later demonstrated the presence of a common mutation of Carnitine Palmitoyl-Transferase II (CPTII). When facing rhabdomyolysis of obscure origin, nephrologists must keep in mind the possibility that even adult patients may have a genetic defect of energy metabolism. In these cases, patients usually experience rhabdomyolysis during exertion, fasting, or infection. CPTII deficiency often has a subtle presentation and might be unrecognized until AKI develops. Therefore, it is important to consider a genetic defect of muscle metabolism even in adult patients when a history of rhabdomyolysis of unclear origin is present.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Nephrologists usually face rhabdomyolysis in the setting of the so-called crush syndrome, in which trauma and/or limb compressions lead to the release of a massive amount of myoglobin, potassium, and intracellular organic acids. This may precipitate acute kidney injury (AKI), hyperkalemia, and metabolic acidosis that can be life-threatening and often require urgent hemodialysis [1, 2]. Other causes of rhabdomyolysis include seizures, delirium tremens, idiopathic inflammatory myopathies, compartment syndromes, and some drugs, such as statins [1]. Anyway, less common causes of rhabdomyolysis may lead to AKI as well, possibly representing a diagnostic challenge even for the expert nephrologist. Among these, inherited metabolic myopathies represent a diagnosis that should be considered also in adult patients with rhabdomyolysis-induced AKI. Oliguria may be the presenting complaint of them and the eventual development of renal impairment poses a threat to their lives, so they should be educated to recognize symptoms and signs of ongoing AKI. We present here such a case, in which a man brought to our attention for oliguric AKI was finally diagnosed with a carnitine palmitoyl-transferase II deficiency, which caused massive rhabdomyolysis upon strenuous physical exertion.
Case presentation
A 41-year-old Caucasian man was admitted to our Emergency Department (ED) following the finding of serum creatinine 8 mg/dl at laboratory tests performed for marked asthenia, nausea, and low urinary output with the appearance of “dark urine”. Eight days before the access to our ED, the patient had taken part to a bicycle race, cycling over 100 Km. He was used to such physical efforts, even though since childhood, he needed to ingest huge amounts of carbohydrates and fluids on the day before and during the effort, to avoid the development of myalgias and muscle stiffness following physical exercise. However, in this last occasion, he had not followed such precautions. On his access to our ED, his vital parameters were normal. Upon catheterization, the patient was oliguric. He reported a weight increase of 3 kg in the last days. Physical examination was remarkable for dependent edema. His blood tests showed serum creatinine levels of 8.7 mg/dl, BUN 107 mg/dl, creatine phosphokinase 1737 IU/l, and myoglobin levels of 2832 ng/ml. Blood pH at arterial blood gas analysis was normal as well as serum electrolytes. Additional blood analyses revealed: Hb 12.7 g/dl, leukocytes 10.200/mm3 with a slight left shift, aspartate aminotransferase 30 IU/l, alanine aminotransferase 201 IU/l, lactate dehydrogenase 349 IU/l, normal bilirubin values, creatine kinase MB isoenzyme 15.1 ng/ml, and CRP 2.29 mg/dl. Troponin I levels were not raised. He underwent an abdominal ultrasound, showing kidneys of normal size, with bilaterally slightly increased cortex width and hypoechoic pyramids. No evidence of calyces dilation nor nephrolithiasis was found. He was given intravenous saline and furosemide and admitted to the nephrology Unit to undergo further investigations. On urinalysis, we found a pH of 5.0, albumin 30 mg/dl, haemoglobin 0.2 mg/dl, and 75 WBCs/μl. Specific gravity of patient’s urine was 1.010. We performed microscopic urinary sediment analysis, showing pigmented granular “muddy-brown” casts and the presence of several tubular cells (3–5/high power field), a finding consistent with ongoing acute tubular necrosis (ATN) secondary to rhabdomyolysis, as suggested by elevated myoglobin and CPK levels (Fig. 1).

Urinary sediment analysis with light microscopy shows the so-called “muddy-brown” cast (×200). Top right: higher magnification (×400)
On thorough history taking our patient referred a childhood history of diffuse muscle pain following prolonged or intense physical exercise. In 1995, while he was serving in the army, he experienced a similar episode of sickness after prolonged marching, for which he had to rest in bed for 2 days to recover. He reported that, after this episode, he was diagnosed as an unspecified myopathy after EMG exam and neurologic evaluation. After this episode, the patient learned how to mitigate these symptoms, by the assumption of a high-carbohydrate diet and an increased water intake on the day before and during physical efforts. He reported also that during his bicycle competitions, he was the only member of the team that had not to urinate during the race, and when finally had to urinate at home, urine output was poor with the appearance of “black” urine. The patient was then treated with intravenous hydration, alkalizing therapy, and diuretics, which yielded a progressive recovery of oliguria and renal function in approximately 1 week. On discharge, blood tests of the patient were almost completely normalized. His BUN was 25.2 mg/dl and creatinine 1.35 mg/dl. Myoglobin and creatine kinase levels had come back to the normal, 43.3 ng/ml and 92 IU/l, respectively. He was discharged with the recommendation of drinking at least 2 L of water per day, avoiding any kind of physical exercise in the following month and avoiding strenuous physical exercise thereafter. After discharge, the patient was referred to the local highly-specialized unit of neuromuscular diseases for further evaluation. A muscular biopsy was performed: histologic examination was unrevealing, but on further enzyme analysis, a Carnitine Palmitoyl-Transferase 2 (CPT2) deficiency [isotype 54.3 pmol/min/mg (normal values 452 ± 160); forward 136 (normal values 367 ± 110)] was found. In addition, the patient underwent genetic analysis and tested homozygous for a common mutation (p.S113L9) in the CPT2 gene, providing us the final diagnosis.
Discussion
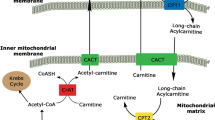
Carnitine Palmitoyl-Transferase (CPT) II deficiency is an autosomal recessive inherited disorder, of which some hundreds cases have been reported at present [3, 4]. Long-chain fatty acids (LFAs) represent the most important form of energy storage in muscle cells in the setting of prolonged exercise and different stress conditions. However, to undergo beta-oxidation, the process in which energy is produced from them, LFAs need to pass from the cytoplasm, in which they are stored, to the inner side of the mitochondrial inner membrane, where the beta-oxidation enzymes are present. Crossing of the mitochondrial membrane is possible only when long-chain fatty acids are trans-esterificated to acylcarnitine, a process enabled by CPT I in the cytoplasm. This acylation allows long-chain fatty acids to cross the mitochondrial membrane, where, on the inner side, are converted back to acyl-CoA by CPT II to start beta-oxidation. Such a process of transport becomes crucial in those conditions in which β-oxidation of long-chain fatty acids is required to meet energy demands in muscles, i.e., during prolonged exercise, fasting, fever, exposure to cold, drugs, and emotional stress [5]. CPT II deficiency is characterized by an abnormal regulation of the CPT II enzyme, whose activity has been shown to be abnormally inhibited when fatty acid metabolism is stressed [6]. Three different phenotypes of the disease have been reported: a lethal neonatal form, a severe infantile hepato-cardio-muscular form, and the most prevalent mild myopathic (“adult”) form [5]. The first signs and symptoms of mild myopathic CPT II deficiency become evident between a mean of 6 and 20 years of age (range 8 months to 50 years) [4]. The clinical picture consists of recurrent attacks of rhabdomyolysis with myalgia, muscle weakness, or stiffness and myoglobinuria, most commonly triggered by exercise or infection; in intervening periods, patients are asymptomatic [7]. Rare but alarming complications may result from rhabdomyolysis, such as respiratory insufficiency, paroxysmal heart arrhythmias, or AKI [8]. However, only few cases of CPT II deficiency leading to AKI have been reported so far [9,10,11,12]. In our case, the patient presumptively had different episodes of AKI before presentation: upon asking, he reported the usual appearance of “black urine” after strenuous physical efforts. Usually, patients learn to manage their symptoms by changing their diet before and during exercise, as our patient did. This has a biochemical rationale: muscles that cannot account on lipid oxidation to produce energy can rely on calories given by carbohydrate consumption to sustain the effort. Our patient decided to seek medical advice this time, because he developed oliguria. Probably, the fact that on the day before the bicycle race, he did not follow the high-carbohydrate diet and he was used to may have precipitated this oliguric episode of AKI. Rhabdomyolysis is not an uncommon cause of AKI [9] and commonly complicates the victims of earthquakes, building collapses and trauma in general. Muscle cell necrosis releases a huge amount of myoglobin into circulation, which is the culprit of rhabdomyolysis-induced AKI [1]. Myoglobin precipitates in combination with Tamm–Horsfall protein in renal tubules, producing the characteristic “muddy-brown” casts that obstruct urine outflow in tubules (Fig. 1). Cast production is also favored by low urinary pH and slow urinary flow: this provides the rationale to aggressively hydrate and alkalinize these patients [2, 8]. AKI is also favored by the toxic effects of iron-induced production of free radicals by the heme moieties of myoglobin that damage epithelial tubular cells [1]. Moreover, the development of a third space in the injured muscles activates the renin–angiotensin–aldosterone and antidiuretic hormone systems which favor oliguria [1, 2]. Apart from the development of AKI, muscle cell necrosis triggers hyperkalemia, hyperphosphatemia, and metabolic acidosis through the release of intracellular electrolytes and organic acids [2]. In the care of these patients, nephrologists have to understand what triggers rhabdomyolysis in the absence of trauma. Most common causes include drugs, muscle hypoxia, limb compression, compartment syndrome, seizures, alcohol, and drug abuse [13] and can be easily identified by careful history taking and physical examination (Table 1). Genetic defects are usually evident as well but when they have milder phenotypes, such as in this case, they may represent a challenge even for the experienced physician. In conclusion, rhabdomyolysis-induced AKI complicates the course of patients with CPT II deficiency [7]. Some of these patients have a mild phenotype and may have a late referral and diagnosis of their condition, so nephrologists must consider this eventuality when treating adult patients with unexplained effort-related rhabdomyolysis with a really suggestive past medical history.
References
Bosch X, Poch E, Grau JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361(1):62–72.
Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19(8):568–74.
Vavlukis A, Eftimov A, Zafirovska P, Caparovska E, Pocesta B, Kedev S, Dimovski AJ. Rhabdomyolysis and cardiomyopathy in a 20-year-old patient with CPT II deficiency. Case Rep Genet. 2014;2014:496410.
Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Asp Med. 2004;25(5–6):495–520.
Joshi PR, Deschauer M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype-phenotype analysis of 50 patients. J Neurol Sci. 2014;338(1–2):107–11.
Lehmann D, Zierz S. Normal protein content but abnormally inhibited enzyme activity in muscle carnitine palmitoyltransferase II deficiency. J Neurol Sci. 2014;339(1–2):183–8.
Nance JR, Mammen AL. Diagnostic evaluation of rhabdomyolysis. Muscle Nerve. 2015;51(6):793:810.
Huerta-Alardìn AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158–69.
Sciacco M, Prelle A, Fagiolari G, Bordoni A, Crimi M, Di Fonzo A, Ciscato P, Lamperti C, D’Adda E, Jann S, Bresolin N, Comi GP, Moggio M. A case of CPT deficiency, homoplasmic mtDNA mutation and ragged red fibers at muscle biopsy. J Neurol Sci. 2005;239(1):21–4.
Topçu Y, Bayram E, Karaoğlu P, Yiş U, Bayram M, Kurul SH. Carnitine palmitoyl transferase II deficiency in an adolescent presenting with rhabdomyolysis and acute renal failure. Pediatr Emerg Care. 2014;30(5):343–4.
Joussain C, Lamireau D, Espil-Taris C, De Précigout V, Vianey-Saban C, Llanas B, Harambat J. A 10-year-old boy with dark urine and acute kidney injury. Pediatr Nephrol. 2011;26(8):1229–33.
Uzel B, Altiparmak MR, Ataman R, Serdengeçti K. Acute renal failure due tocarnitine palmitoyltransferase II deficiency. Neth J Med. 2003;61(12):417–20.
Torres PA, Helmstetter JA, Kaye AM, Kaye AD. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner J. 2015;15(1):58–69.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors have declared no competing interest.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from the patient for the publication of his case.
About this article

Cite this article
Cucchiari, D., Colombo, I., Amato, O. et al. Exertional rhabdomyolysis leading to acute kidney injury: when genetic defects are diagnosed in adult life. CEN Case Rep 7, 62–65 (2018). https://doi.org/10.1007/s13730-017-0292-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-017-0292-z