
Abstract
The genetic structure of the Iberian honey bee (Apis mellifera iberiensis) was studied by analysing 10 microsatellite loci in 362 workers representative of nine Spanish provinces. Heterozygosity values of Iberian honeybee populations are intermediate between African and west European ones whereas allelic diversity is remarkably high at several loci. There is no definite geographic structure of Iberian honeybee populations. At a peninsular scale, the expected clinal pattern observed with mitochondrial data has been probably lost due to the extensive practice of mobile beekeeping and increased colony trade-off. Due to these practices, it is expected that the genetic homogenisation will increase during the next years. Though this might have positive effects on honey production, putative ecotypes existing in Iberia would be prone to disappear.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Mediterranean populations of the honeybee Apis mellifera belong to four evolutionary lineages characterised by both morphological (Ruttner 1988) and molecular characters (Smith et al. 1991; Garnery et al. 1992; Estoup et al. 1995; Arias and Sheppard 1996; Franck et al. 2001). Each of them has a particular distribution: Africa (A lineage), West and North Europe (M lineage), Southeast Europe (C lineage) and Near and Middle East (O lineage). Lineages A and M intergrade in the Iberian Peninsula giving rise to the subspecies Apis mellifera iberiensis Engel, 1999. Hybridisation in this subspecies is observed in morphological and ethological characters (Ruttner 1988).
The hybrid origin of the Iberian honeybee can be observed in the mitochondrial DNA as it shows haplotypes belonging to both A and M evolutionary lineages what has been fully documented in thousands of samples representative of the whole peninsula (Miguel et al. 2007; Cánovas et al. 2008). Honeybee populations from the Iberian Peninsula show a definite structure with regard to mitochondrial markers, as the frequency of African haplotypes decreases from southwestern to northeastern Iberia while West European haplotypes increases in the same ratio. This result is likely supported by both historical and selective factors: successive cycles of colonisation, extinction, survival on refugia and recovery of Iberian populations during Pleistocene glaciation events, and adaptation to local conditions (Miguel et al. 2007; Cánovas et al. 2008).
It is unclear why the clinal variation found in Iberia for mtDNA haplotypes does not occur with regard to microsatellite markers. First results reported by Garnery et al. (1998b) showed that microsatellite loci are much less variable in Iberia and France than in Africa, as a result of a hypothetical bottleneck in Iberian populations during the last glaciation (Franck et al. 1998; Garnery et al. 1998b). Microsatellites indicate a clear-cut disruption between Africa and Europe. Moreover, the genetic profiles are rather homogeneous from Spain to Scandinavia (Garnery et al. 1998b). Later results suggested a more complex situation as genetic diversity detected in populations from Murcia (De la Rúa et al. 2002b), Balearic Islands (De la Rúa et al. 2003) and Andalusia (De la Rúa et al. 2004b) showed intermediate values between African and West European populations. However, Miguel et al. (2007) found that nuclear genetic diversity was similar between Iberian, French and Belgian populations, and hypothesised that this similarity was due to higher mutation rates of the microsatellites, which would rapidly increase the initial diversity of refugee populations after the last glaciation maximum, thus masking the effects of the re-colonisation process in a relatively short time. These conclusions were based on samples coming from 18 Iberian populations covering a large peninsular area but excluding the northwestern quadrant.
Further, it should be noted that due to the nature of nuclear DNA and the mating behaviour of honeybees, the geographic distribution of microsatellite markers in Iberian populations could have been changed by apiculture practices during the last 20 years. Migratory beekeeping has become an extended practice in all Mediterranean countries, being fully developed in the Iberian Peninsula (Ruíz and Ruíz 1986). In Spain, 80% of the estimated number of colonies is moved in a yearly cycle that often includes three movements from winter to spring and later on to summer–fall locations. This activity permits optimum exploitation of flowering periods in a latitudinal and altitudinal gradient but originates a potential bi-directional genetic flow between mobile and resident hives (Perrier et al. 2003). Furthermore, honeybee populations have been decimated by the parasitic mite Varroa destructor that killed thousands of Iberian colonies during the 1980s and the 1990s (Gómez-Pajuelo 1988; Gómez-Pajuelo 2003). The genetic changes in local honeybee populations have possibly been speed up by the recovery of these colonies, as many beekeepers purchased colonies from other regions, thus favouring the homogenization of Iberian honeybees at a peninsular scale.
The initial hypothesis to be tested postulates that Iberian honeybee populations will show a decreasing diversity from south to north (according to the general pattern found between Africa and Europe), and an isolation-by-distance effect as a main factor to explain the differences between populations (according to Miguel et al. 2007). To this aim, 10 microsatellite markers were studied in nine populations of the Iberian Peninsula, with particular attention to those located at the northwestern quadrant.
2 Materials and methods
2.1 Sampling
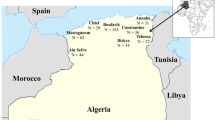
A total of 362 honeybee workers (one worker bee per colony) were collected from the inner frames of the beehive. Samples were from 49 apiaries located in nine Spanish provinces with different beekeeping practices (Table I and Figure 1; detailed in Appendix I). In that sense, Cáceres, Cádiz, Tarragona and Valencia hold predominantly migratory colonies while the other provinces hold predominantly stationary colonies. Samples were grouped at a province scale, according to the criterion of the maximum radius of mating flight. Drones and queens of different colonies gather in a drone congregation area (DCA) and thus localities with shared DCAs have a panmictic population structure. Recent conservation studies aimed to identify pure breed mating areas estimated 20 km as a minimum distance for genetic isolation (Kraus 2005). Samples overlapping circles of 20 km radius were considered to make a single population (Figure 1); for this reason, samples of the same province were equated to a single population and are named in this work according to the corresponding Spanish name.

Iberian honeybee populations analysed by microsatellite loci. The area including the points shows the minimum distance of genetic isolation and refers to a panmictic population structure (Kraus 2005). (Ast Asturias, Bur Burgos, Cac Cáceres, Cad Cádiz, Cor Coruña, Ore Orense, Pon Pontevedra, Tar Tarragona, Val Valencia).ithe geographical variation for: a the first component of the AFC, which accounted for 26.30% of the total allele variation in Apis mellifera across Iberian Peninsula and b the second component, which accounted for 45.80% cummulative variation. First component follows a gradient that peaks at the east and it is probably correlated with areas of intensive apiculture practices. The second component shows the high differentiation of samples from Burgos. The tlikely genetic boundaries suggested by the Monmonier’s algorithm with width proportional to intensity were represented as solid lines (Manni et al. 2004).
2.2 Microsatellite amplification
Samples were brought to the laboratory preserved in absolute ethanol. Total DNA was extracted from one leg following Walsh et al. (1991) with a Chelex 5% based protocol. Honeybee populations were scored using 10 microsatellite loci: A007, A024, A028, A088, Ap043, Ap055, Ap066, Ap081, A107 and A113 (Solignac et al. 2003). One primer of each pair was labelled with fluorescence at the 5′ end (Applied Biosystems, Foster City, CA, USA) and the PCR reactions were carried out following Solignac et al. (2003). Allele sizes were determined in an ABI PRISM 3700 DNA Sequencer (Applied Biosystems) and scored using STRand v2.3.106 (Pereira et al. 2007).
2.3 Genetic variability analysis
Unbiased estimates and standard deviations of gene diversity of microsatellite loci (H e) were calculated following Nei (1978). Genotypic linkage disequilibrium and genic and genotypic differentiation were also computed. Probabilities of random departure from Hardy–Weinberg equilibrium and inbreeding coefficient (F IS) were calculated using the exact test method (Raymond and Rousset 1995a) and the permutation test (Belkhir et al. 2004). Data were analysed using the GENEPOP ON THE WEB (http://genepop.curtin.edu.au/, Raymond and Rousset 1995b) and GENETIX v4.0.5 package (Belkhir et al. 2004). FreeNA software v1 was used to infer the presence of null alleles that lead to the overestimation of both F ST and genetic distance in cases of significant population differentiation (Chapuis and Estoup 2007).
These indices do not allow for a statistical comparison of alleles richness for populations with different sample sizes. Using rarefaction method, the expected number of alleles in a random subsample from a population with an observed allele richness distribution can be calculated (Kumar et al. 1999). This process yielded after 200 iterations, the mean number of sampled individuals required to detect all the distinct alleles within a population.
2.4 Population structure analysis
Population structure was inferred using STRUCTURE v2.3 software by the method of Pritchard et al. (2000) from multilocus genotype data. We assumed a model in which there were from 1 to 12 populations (K clusters). Each K was replicated 20 times for 100,000 iterations after a burn-in period of 50,000, without any prior information on the population of origin of each sampled individual.
Population substructure was evaluated by calculating pairwise and global F ST values (Wright 1951); Cavalli-Sforza and Edwards (1967) distance was calculated using POPULATIONS software v1.2.30 over 10,000 bootstraps on individuals (http://bioinformatics.org/~tryphon/populations/). Hypotheses of isolation by distance were tested according to Slatkin (1993) with a Mantel test (Mantel 1967). Probabilities were read directly from the distribution of 1,000 randomised matrices computed by permutation. Mantel’s test was performed using the procedure of the R statistical software (R Development Core Team 2008).
Spatial structure of genetic variation within populations, an important interacting influence on evolutionary processes, can be analysed in detail by assessing associations between the genetic relatedness of pairs of individuals and the geographical distance (Epperson and Li 1996). Multiallelic and multilocus approaches were performed to estimate spatial autocorrelation from genetic distances and mean number of common alleles (Smouse and Peakall 1999). Geographic distances were grouped in 20 categories, including at least 1,200 comparisons per category. A Monte Carlo simulation was performed to estimate the deviation from the random distribution with a 95% confidence interval (Streiff et al. 1998).
Correlation analyses fail to identify the existence of genetic barriers, namely, the areas where a given variable shows an abrupt rate of change. Monmonier’s maximum difference algorithm enables a better interpretation of microevolutionary processes, such as gene flow, genetic drift, and selection (Manni et al. 2004). Cavalli-Sforza and Edwards (1967) genetic distances and F ST applied to pairs of provinces were used to produce the genetic matrices. Monmonier’s maximum difference algorithm was then used to identify putative genetic boundaries across the Iberian Peninsula by BARRIER v2.2 software (Manni et al. 2004).
A factorial analysis of correspondence (AFC) using the individual microsatellite allelic composition was performed (Belkhir et al. 2004). Ordination summarises all the variation in the study area and accommodates each population as a study unit (Manel et al. 2003). Then, the inverse distance weighted interpolation of the major principal components leads to a synthesis map, which gives insights into the spatial pattern concerned (Hanotte et al. 2002). AFC analyses were performed using GENETIX and interpolation using GRASS GIS software (GRASS Development Team 2008).
3 Results
3.1 Genetic variability
Rarefaction curves for total allele richness in A. m. iberiensis showed that the minimum sample size for 95% of the expected total allele richness was attained in all A. m. iberiensis populations.
Allele frequencies for all loci per sampling site combinations are detailed in Appendix II, including observed (H o) and expected heterozygosity (H e). In total, 124 alleles were detected and all loci were polymorphic in almost all populations, except Cádiz, Asturias and Cáceres for locus A028. The mean number of alleles in each population varied between 5 (La Coruña) and 7.5 (Valencia, Table I). The value observed in La Coruña might be due to the small sample size (n = 18), although the total number of alleles (five alleles) was similar to that found in other populations with higher sample sizes (e.g., Orense, Pontevedra or Cádiz).
Tarragona, Valencia and Asturias showed a positive and significant value of F IS (Table I), and also a significant departure of Hardy–Weinberg equilibrium due to heterozygote deficiency. Samples from Cádiz also showed a significant departure of H-W equilibrium for exact test values but F IS values did not indicate a heterozygote excess. The other populations showed a situation close to Hardy–Weinberg equilibrium.
Multiple probability tests for all populations detected signs of linkage disequilibrium between loci A007 and A24, and A028 and Ap043 (P > 0.05), although there were only single significant values for every single population. Genic and genotypic differentiation tests demonstrated the genetic differentiation existing among pairs of populations except for La Coruña, Orense and Pontevedra with Cáceres, and Orense with La Coruña (Table II).
3.2 Population structuring
The populations of A. m. iberiensis did not show a definite structure as shown by the Bayesian approach. This analysis suggests a K = 1 best describing the situation among samples. Accordingly, the global F ST (0.036) and the mean genetic differentiation (F ST) of pair wise comparisons were low, ranging from 0.101 (Burgos and Cádiz) to 0.006 (Orense and Cáceres; Table II). The values observed between Orense- Pontevedra (these two provinces are located at the north-western side of the Iberian Peninsula), and Valencia–Tarragona (located at the eastern Mediterranean coast) were not significant.
The absence of a geographical pattern was confirmed by the non-significant results (Mantel test) between geographic and Cavalli-Sforza and Edwards distances (r = 0.233; P = 0.141), although well-supported groups were observed in the genetic distances dendrogram (Figure 2). Tarragona and Valencia were differentiated from the rest of populations, and two groups in which populations from North and South Iberia are mixed (Cáceres and Orense, and Asturias and Cádiz) were formed.

Neighbour-joining unrooted tree produced from Cavalli-Sforza and Edwards (1967) distances between Iberian honeybee localities.
Spatial autocorrelation showed that geographical distance is not the main determinant factor of spatial genetic structure in the Iberian honeybee populations (Figure 3). Samples separated less than 200 km showed significant higher numbers of common alleles and smaller genetic distances between individuals, but similar values were also found at distances higher than 650–750 km. The main contribution to this distance class was the relation between samples from Asturias and Cádiz. Distances between samples close to 400 km, showed significant genetic similarity values too, but lower than the other categories. In this case, the higher contribution was the relationship between Burgos with La Coruña and Pontevedra, and Cáceres with Cádiz.

Correlograms plot: a genetic distances and b mean number of common alleles as a function of geographic distances. The 95% confidence interval (dashed lines) are also shown. The number of pairwise comparisons within each distance class represented as a dot is presented below the plotted lines. Note that significant genetic similarities were shown between samples separated several hundred kilometres.
Two main genetic boundaries were detected by the Monmonier’s algorithm. The most significant was detected between the populations from Tarragona and Valencia and the other Iberian honeybee populations (Figure 1). This result was corroborated by the correspondence analysis. The first component showed a gradient between eastern (darkest areas) and the rest of Iberian samples, explaining 26.30% of the total microsatellite allele variation (Figure 1). The extreme of that gradient (lightest areas in Figure 1) showed the similarities between Cádiz and northwestern localities. The second genetic barrier was found between Burgos and the surrounding populations. The second component of the ordination showed the same pattern, adding 19.50% to the cumulative variation of 45.80%. In this case, the extreme of the gradient (darkest areas) was made up by the similarities of western Iberian populations.
4 Discussion
4.1 Genetic diversity and structure of Iberian honeybees
The number of alleles per locus and the heterozygosity values found in this study are higher than those reported by Franck et al. (1998) for three samples from Sevilla, Segovia and San Sebastián (H e = 0.283–0.331), and are more similar to those reported by De la Rúa et al. (2002b) for Murcia (SE Spain; H e = 0.448) and by Miguel et al. (2007) for different Iberian regions (H e = 0.442–0.516). As a whole, the sampled populations are not structured according to a definite geographic pattern, in contrast to that found in mitochondrial haplotypes (Cánovas et al. 2008). This finding does not give support to our initial hypothesis stating that populations from South Spain will show higher allelic diversity than those from North Spain due to the influence of African populations at the South. Franck et al. (1998) reported a clear trend towards decreasing microsatellites diversity from Tropical Africa to northwest Europe, that agrees with the recent hypothesis of an African origin for A. mellifera based on the study of single-nucleotide polymorphisms (SNPs; Whitfield et al. 2006). At this large geographic scale, Iberian populations show congruent intermediate values between African populations—which have the highest level of variability-, and west European populations north of the Pyrenees—which have the lowest (Franck et al. 2001). However, the pattern seems to have disappeared within the Iberian Peninsula. Only the Pyrenees seems to be a barrier between populations at each side (Miguel et al. 2007). Although Miguel et al. (2007) concluded that ‘the shorter the geographic distance between populations, the greater their genetic similarity’, our data exemplify too many exceptions. Possibly, the validity of that conclusion depends on the degree of change experimented by the populations subjected to comparison due to beekeeping activities.
Morphology, allozyme and mtDNA data clearly show a wide hybridisation area between African and European lineages where A. m. iberiensis occur (Ruttner 1988; Garnery et al. 1998a; Franck et al. 1998; Arias et al. 2006; Miguel et al. 2007; Cánovas et al. 2008). Data from SNPs led to postulate the African origin of A. mellifera and that there were at least two subsequent expansions into Eurasia—a western expansion into Europe (M) and one or more eastern expansion into Asia and Europe (O and C; Whitfield et al. 2006). The intermediate values of microsatellite heterozygosity agree with the survival of Iberian populations during the last glaciation event—which lately colonised Western Europe during present postglacial period—and the arrival of the hypothesised colonisation waves from North Africa (Garnery et al. 1998b; Franck et al. 1998; Miguel et al. 2007; data presented here).
4.2 Genetic consequences of beekeeping practices
In spite of the lack of a geographic structure at a peninsular scale showed by the STRUCTURE results, signs of a genetic substructure were found. Populations of the Mediterranean region, Tarragona and Valencia, showed deviations of the Hardy–Weinberg equilibrium which indicate a deficiency of heterozygotes congruent with FIS values. The group made by these two geographically close populations is supported by the genetic distances and the genetic boundary found after the spatial interpretation of the ordination analysis (Figure 1). The regional laws that have compelled the beekeepers from Valencia to periodically move their colonies 5 km away from citrus areas during the flowering period (from April until the end of May), in order to avoid cross-pollination that produces citrus fruits with seeds, may have influenced the genetic variability of these colonies (De la Rúa et al. 2004a).
The marked degree of homogenisation at a peninsular scale suggests that a large amount of gene flow has occurred between Iberian populations in recent times. Two processes seem to account for most of these findings. The first is related to the need of a rapid recovery of populations affected by Varroa. A large amount of honeybee colonies of the Iberian Peninsula were destroyed in the 1980s by the parasitic mite V. destructor (Gómez-Pajuelo 1988 and personal communication), with losses up to 50% in some cases. Population recovery was achieved either by swarming (or partition of surviving colonies) and by purchasing colonies (or mated queens) often from distant localities. Unfortunately, there are few documented data about the intensity of this process and only oral reports from beekeepers can be used as evidence of this process.
The second process is mobile beekeeping that affects 28.58% of the Spanish beeyards (ranging from 94.86% of the registered colonies in Valencia to 0.98% in Galicia, including all the provinces: La Coruña, Pontevedra and Orense; data from 2009; http://www.mapa.es/app/Miel/indicadores/Indicadores.aspx?lng=es, accessed 12/02/2010). Mobile beekeeping allows for a bidirectional gene flow between mobile and local colonies though as noted by Perrier et al. (2003), it seems that there is a skewed flow from local to mobile populations. Beekeeping practices vary between provinces as stated before though there is a general trend to move the colonies from areas with a Mediterranean climate (and an early flowering season), to areas with a milder summer and late flowering period (mountains of central and north Spain). This means that few areas of Spain can avoid the influence of mobile beekeeping.
The values of genetic diversity, genetic differentiation and H–W equilibrium found in Burgos population (central–northern Spain) are likely related to this process. Since the last decade there are in this province summer settlements of colonies coming either from south and central Spain or from the Basque Country, thus favouring the gene flow between geographically distant populations. On the contrary, Asturias shows lower heterozigosity than other northern regions but an H–W disequilibrium with a heterozigosity excess. These results could show the relative isolation of this area from the most frequent paths of mobile beekeeping that particularly affect more southern regions and an enhanced practice of swarming to recover lost colonies, preventing homogenization and increasing heterozigosity. In provinces close to Asturias located in Galicia (La Coruña, Pontevedra, Orense), mobile beekeeping is not intense but heterozigosity levels increase probably due to the sharp transition of African and European lineages (Cánovas et al. 2008), that favours hybridization between both lineages.
Historical and climatic factors would produce a natural distribution pattern of mtDNA haplotypes across the Iberian Peninsula that remains stable due to the maternal inheritance of mtDNA and the mating behaviour of honeybees. Nevertheless, anthropogenic factors may rapidly change this pattern because of the great development of mobile beekeeping and man-made replacement of native colonies (Cánovas et al. 2008), as it was found on the islands of Formentera and Ibiza (De la Rúa et al. 2001) and on the Canary Islands (De la Rúa et al. 2002a). It should be noted that the geographic distribution of microsatellite markers in Iberian populations has been possibly changed by apiculture practices during the last 20 years. It can be concluded that in relation to the nuclear genetic diversity reflected by microsatellites, the honeybee populations of Iberia make up an almost unique population, with a discrete differentiation between an eastern Mediterranean subpopulation (represented by Tarragona and Valencia) and a western subpopulation including the other peninsular areas. This unexpected level of homogenisation is likely caused by a trade-off of colonies from distant sources and by mobile beekeeping—the importance of each of these two processes must be ascertained. The overall effect of both processes is the increase of the genetic diversity of colonies, what is thought to increase the productivity and fitness of the colonies (Mattila and Seeley 2007). However, this homogenisation could also lead to the loss of putative ecotypes, that is, local populations well adapted to particular conditions after many generations of selection by natural and man-made factors. An effort to describe and preserve these ecotypes is thus a strong commitment for the next years.
References
Arias, M.C., Sheppard, W.S. (1996) Molecular phylogenetics of honeybee subspecies (Apis mellifera L.) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 5, 557–566
Arias, M.C., Rinderer, T.E., Sheppard, W.S. (2006) Further characterization of honey bees from the Iberian Peninsula by allozyme, morphometric and mtDNA haplotype analyses. J. Apic. Res. 45, 188–196
Belkhir, K., Borsa, P., Chikhi, L., Raufaste, N., Bonhomme, F. (2004) GENETIX, version: 4.05.2: logiciel sous WindowsTM pour la génétique des populations. Laboratoire Génome, Populations, Interactions. Université de Monftpellier II. Available from: http://www.genetix.univ-montp2.fr/genetix/genetix.htm
Cánovas, F., De la Rúa, P., Serrano, J., Galián, J. (2008) Geographic patterns of mitochondrial DNA variation in Apis mellifera iberiensis (Hymenoptera: Apidae). J. Zool. Syst. Evol. Res. 46, 24–30
Cavalli-Sforza, L.L., Edwards, A.W.F. (1967) Phylogenetic analysis: models and estimation procedures. Evolution 32, 550–570
Chapuis, M.P., Estoup, A. (2007) Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631
De la Rúa, P., Galián, J., Serrano, J., Moritz, R.F.A. (2001) Molecular characterization and population structure of the honeybees from the Balearic Islands (Spain). Apidologie 32, 417–427
De la Rúa, P., Serrano, J., Galián, J. (2002a) Biodiversity of Apis mellifera populations from Tenerife (Canary Islands) and hybridisation with East European races. Biod. Cons. 11, 59–67
De la Rúa, P., Serrano, J., Galián, J. (2002b) Biodiversity of Apis mellifera populations from Tenerife (Canary Islands) and hybridisation with East European races. Biod. Cons. 11, 59–67
De la Rúa, P., Galián, J., Serrano J., Moritz R.F.A. (2002b) Microsatellite analysis of non-migratory colonies of Apis mellifera iberica from south-eastern Spain. J. Zool. Syst. Evol. Res. 40, 164–168
De la Rúa, P., Galián, J., Serrano, J., Moritz, R.F.A. (2003) Genetic structure of Balearic honeybee populations based on microsatellite polymorphism, Genet. Sel. Evol. 35, 1–12
De la Rúa, P., Jiménez, Y., Galián, J., Serrano, J. (2004a) Evaluation of the biodiversity of honey bee (Apis mellifera) populations from eastern Spain. J. Apic. Res. 43, 162–166
De la Rúa, P., Hernández-García, R., Pedersen, B.V., Galián, J., Serrano, J. (2004b) Molecular diversity of honeybee Apis mellifera iberica L (Hymenoptera: Apidae) from western Andalusia. Arch. Zootec. 53, 195–203
R Development Core Team (2008) R: A language and environment for statistical computing v2.8.1. Available from: http://www.r-project.org.
GRASS Development Team (2008) Geographic resources analysis support system (GRASS) v6.3.0. Available from: http://grass.osgeo.org
Epperson, B., Li, T. (1996) Measurement of genetic structure within populations using Moran's spatial autocorrelation statistics. Proc. Natl. Acad. Sci. USA 93, 10528–10532
Estoup, A., Garnery, L., Solignac, M., Cornuet, J.M. (1995) Microsatellite variation in honey bee (Apis mellifera L.) populations: hierarchical genetic structure and test of the infinite allele and stepwise mutation models. Genetics 140, 679–695
Franck, P., Garnery, L., Solignac, M., Cornuet, J.M. (1998) The origin of West European subspecies of honeybees (Apis mellifera): new insights from microsatellite and mitochondrial data. Evolution 52, 1119–1134
Franck, P., Garnery, L., Loiseau, A., Hepburn, H.R., Solignac, M., Cornuet, J.M. (2001) Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity 86, 420–430
Garnery, L., Cornuet, J.M., Solignac, M. (1992) Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol. Ecol. 1, 145–154
Garnery, L., Frank, P., Baudry, E., Vautrin, D., Cornuet, J.M., Solignac, M. (1998a) Genetic diversity of the west European honeybee (Apis mellifera mellifera and A. m. iberica). I. Mitochondrial DNA. Genet. Sel. Evol. 30, 31–47
Garnery, L., Frank, P., Baudry, E., Vautrin, D., Cornuet, J.M., Solignac, M. (1998b) Genetic diversity of the west European honeybee (Apis mellifera mellifera and A. m. iberica). II. Microsatellite loci. Genet. Sel. Evol. 30, 49–74
Gómez-Pajuelo, A. (1988) Situation of the varroatosis in Spain and Portugal. Present status of varroatosis in Europe and progress in the Varroa mite control, in: Cavalloro, R. (ed.) Proceedings of the Meeting of EC Experts’ Group Udine, Italy, pp. 41–43. ommission of the European Communities, Luxembourg
Gómez-Pajuelo, A. (2003) Introducción de reinas en las colmenas, Vida Apícola117
Hanotte, O., Bradle, D.G., Ochieng, J.W., Verjee., Y, Hill, E.W., Rege, E.O. (2002) African pastoralism: genetic imprints of origins and migrations. Science 296, 336–339
Kraus, F.B. (2005) Requirements for local population conservation and breeding. In: Lodesani, M., Costa, C. (eds.) Beekeeping and conserving biodiversity of honeybees. Sustainable bee breeding, theoretical and practical guide, pp. 87–107. Northern Bee Books, Hebden Bridge
Kumar, J., Nelson, R.J., Zeigler, R.S. (1999) Population structure and dynamics of Magnaporthe grisea in the Indian Himalayas. Genetics 152, 971–984
Manel, S., Schwartz, M.K., Luikart, G., Tarberlet, P. (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol. Evol. 18, 189–197
Manni, F., Guérard, E., Heyer, E. (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by “Monmonier’s algorithm”. Hum. Biol. 76, 173–190
Mantel, N. (1967) The detection of disease clustering and a generalised regression approach. Cancer Res. 27, 209–220
Mattila, H.R., Seeley, T.D. (2007) Genetic diversity in honey bee colonies enhances productivity and fitness. Science 317, 362–364
Miguel, I., Iriondo, M., Garnery, L., Sheppard, W.S., Estonba, A. (2007) Gene flow within the M evolutionary lineage of Apis mellifera: role of the Pyrenees, isolation by distance and post-glacial re-colonization routes in the western Europe. Apidologie 38, 141–155
Nei, M. (1978) Estimation of average heterozigosity and genetic distance from a small number of individuals. Genetics 89, 583–590
Pereira, T.C., Bittencourt, V.D.A., Secolin, R., de Souza-Rocha, C., de Godoy- Maia, C., Lopes-Cendes, I. (2007) Strand analysis, a free online program for the computational identification of the best RNA interference (RNAi) targets based on Gibbs free energy. Genet. Mol. Biol. 30, 1206–1208
Perrier, C., Strange, J., Langella, O., Sheppard, W.S., Garnery, L. (2003) Diversité génétique, introgressions mitochondriales et nucléaires dans une population d'abeilles des Landes de Gascogne. Actes du Bureau de Recherches Génétiques 4, 79–100
Pritchard, J., Stephens, M., Donnelly, P. (2000) Inference of population structure using multilocus genotype data. Genetics 155, 945–959
Raymond, M., Rousset, F. (1995a) An exact test for population differentiation. Evolution 49, 1280–1283
Raymond, M., Rousset, F. (1995b) GENEPOP version 1.2: a population genetics software for exact test and ecumenicism. J. Hered. 86, 248–249
Ruíz, M., Ruíz, J.P. (1986) Ecological history of trashumance in Spain. Biol. Conserv. 37, 73–86
Ruttner, F. (1988) Biogeography and taxonomy of honeybees. Springer-Verlag, Berlin
Slatkin, M. (1993) Isolation by distance in equilibrium and nonequilibrium populations. Evolution 47, 264–279
Smith, D.R., Palopoli, M.F., Taylor, B.R., Garnery, L., Cornuet, J.M., Solignac, M., Brown, W.M. (1991) Geographical overlap of two mitochondrial genomes in Spanish honeybees (Apis mellifera iberica). J. Hered. 82, 96–100
Smouse, P.E., Peakall, R. (1999) Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82, 561–573
Solignac, M., Vautrin, D., Loiseau, A., Mougel, F., Baudry, E., Estoup, A., Garnery, L., Haberl, M., Cornuet, J.M. (2003) Five hundred and fifty microsatellite markers for the study of the honeybee (Apis mellifera L.) genome. Mol. Ecol. Notes 3, 307–311
Streiff, R., Labbe, T., Bacilieri, R., Steinkellner, H., Glossl, J., Krener, A. (1998) Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol. Ecol. 7, 317–328
Walsh, P.S., Metzger, D.A., Higuchi, R. (1991) ChelexR 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10, 506–513
Whitfield, C.W., Behura, S.K., Berlocher, S.H., Clark, A.G., Johnston, J.S., Sheppard, W.S., Smith, D.R., Suarez, A.V., Weaver, D., Tsutsui, N.D. (2006) Thrice out of Africa: ancient and recent expansions of the honey bee, Apis mellifera. Science 314, 642–645
Wright, S. (1951) The genetical structure of populations. Ann. Eugen. 15, 323–354
Acknowledgements
Thanks are due to the many beekeepers and associations who generously collaborated with us in collecting the samples. Special thanks to O Rodríguez, J Ornia, E Muñiz, U Mediel and COAG association. Thanks to Dr. O Langella for his support with Populations software. We thank three anonymous referees for their valuable suggestions. This research has been supported by projects RZ00-013 (Instituto Nacional de Investigaciones Agrarias, INIA, Spain), EVK2 2000–00628 (European 5th Framework), BOS2003-9765 (Ministerio de Educación y Ciencia, Spain) and API06-010 COORD-2 (Ministerio de Agricultura, Pesca y Alimentación, Spain).
La variabilité des microsatellites révèle les effets de l’apiculture sur les populations d’abeilles espagnoles.
Apis mellifera / génétique des populations / cartes synthétiques
Zusammenfassung – Die Variabilität von Mikrosatelliten zeigt den Einfluss der Imkerei auf Populationen der Honigbiene in Spanien. Es ist bisher nicht klar, ob der bei den Bienen Iberiens beobachtete, den Breitengraden folgende Gradient der mitochondrialen DNA auch bei Mikrosatellitenmarkern auftritt. Erste Ergebnisse dazu zeigten eine höhere Variabilität von Mikrosatelliten in Afrika gegenüber Iberien und Frankreich, als Folge eines vermuteten genetischen Flaschenhalses während der letzten Eiszeit. Neuere Ergebnisse zeigten jedoch eine vergleichbar große Diversität in Populationen aus Belgien, Frankreich und Iberien. Es wurde vermutet, dass diese Ähnlichkeit durch die höheren Mutationsraten der Mikrosatelliten verursacht wurde, die die genetischen Effekte der Wiederbesiedlung nach der letzten Eiszeit verdeckt haben könnten.
Die zu testende Ausgangshypothese besagt, dass iberische Honigbienenpopulationen eine abnehmende Diversität von Süden nach Norden zeigen, wobei die Unterschiede zwischen Populationen hauptsächlich durch einen Isolation-by-Distance-Effekt zu erklären wären. Dazu wurden 10 Mikrosatellitenmarker in neun Populationen der Iberischen Halbinsel untersucht.
Der ausgeprägte Grad an genetischer Homogenität lässt vermuten, dass in jüngster Zeit ein beträchtlicher Genfluss zwischen den verschiedenen iberischen Populationen stattgefunden hat. Zwei verschiedene Prozesse können dafür verantwortlich sein. Der eine hängt damit zusammen, dass durch Varroa verlorenen gegangene Bienenvölkern wieder aufgebaut werden mussten. Dazu wurden verstärkt Ableger oder Schwärme gebildet, sowie Völker (oder begattete Königinnen), oft aus entfernten Gegenden, zusätzlich gekauft. Der andere Einfluss ist die Wanderimkerei, die im Durchschnitt 28.58% der spanischen Bienenstände betrifft (von 94.86% in Valencia bis 0.98% in Galizien, einschließlich La Coruña, Pontevedra und Orense).
Aus den Ergebnissen schließen wir, dass die Honigbienen von Iberien eine nahezu homogene Population bilden, jedoch mit einer Differenzierung zwischen einer östlichen, mediterranen Subpopulation (repräsentiert von Tarragona und Valencia), und einer westlichen, die die anderen Gebiete der Halbinsel einschließt.
Dieser unerwartete Grad an Homogenisierung wird wahrscheinlich durch Handel mit Völkern zwischen entfernten Gegenden und durch Wanderimkerei verursacht – das Ausmaß dieser Einflüsse muss jedoch noch bestimmt werden. Der Gesamteffekt dieser Prozesse liegt in einer Zunahme der genetischen Diversität innerhalb der Völker, zur Steigerung der Fitness und der Produktivität. Jedoch wird diese Homogenisierung zum Verlust möglicherweise vorhandener Ökotypen beitragen, das heißt, von Populationen, die an spezifische lokale Bedingungen nach vielen Generationen natürlicher und imkerlicher Selektion gut angepasst sind. In den nächsten Jahren wird es eine wichtige Aufgaben sein, Beiträge zur Erhaltung dieser Ökotypen zu leisten.
Apis mellifera / Populationsgenetik / Synthesekarten
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript editor: Marina Meixner
Appendices
Appendix I
Appendix II
Rights and permissions
About this article
Cite this article
Cánovas, F., de la Rúa, P., Serrano, J. et al. Microsatellite variability reveals beekeeping influences on Iberian honeybee populations. Apidologie 42, 235–251 (2011). https://doi.org/10.1007/s13592-011-0020-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-011-0020-1