
Abstract
Beekeeping as a staple of agriculture in Kazakhstan is believed to have emerged when external bee colonies were introduced into the country. To date, six subspecies have been distributed across commercial apiaries (Apis mellifera sossimai, A. m. carpatica, A. m. mellifera, A. m. ligustica, A. m. caucasica, and A. m. carnica). In this study, we analyzed mitochondrial DNA from 21 geographically distinct populations with diverse subspecies composition and discovered 19 unique haplotypes belonging to C and M evolutionary lineages. Analysis of 9 microsatellite loci revealed strong polymorphism, with a range of 5 to 35 alleles at each locus. The observed heterozygosity across the populations varied from 0.2892 to 0.9099, and clustering analysis only allowed differentiation of each population based on its geographical location. High degree of admixture among subspecies was identified in every population, including a potential introgression from the endemic bee A. m. pomonella. Unregulated cross-breeding for the past 50 years has laid the foundation for the promiscuous genetic nature of honeybee populations in Kazakhstan.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Apis mellifera L., or the western honeybee, is a species widely distributed on all continents except Antarctica. Apis mellifera L. can be divided into five evolutionary lineages (A(+ Z), M, C, O, Y) (Alburaki et al. 2011; Ilyasov et al. 2020) and consists of 27–30 subspecies according to morphological and phylogeographic research (Ruttner 1988; Whitfield et al. 2006). Such a large number of subspecies was formed due to the isolation and subsequent accumulation of genetic differences. The intensive hybridization of the subspecies is currently still ongoing (Dogantzis and Zayed 2019; Ilyasov and Kwon 2021).
Several hypotheses based on morphological and DNA analysis data have been proposed to explain the origins of the subspecies and diversity of evolutionary lineages. Both Africa (Whitfield et al. 2006) and the Middle East (Cridland et al. 2017; Ji 2021) have been considered possible centers for the origin of A. mellifera L., with a subsequent bee migration to Eurasia via the Iberian and Arabian peninsulas. This remains a subject of active debate, and more recent studies prioritize West Asia or North Africa as points of origin (Tihelka et al. 2020; Dogantzis et al. 2021).
The territory of Kazakhstan is vast and covers various types of natural conditions, including areas suitable for beekeeping. Archeological studies of historical monuments in Altai (Kazakhstan side) and Siberia, as well as research into the history of Altai languages suggest that bees inhabited the territory of present-day Altai in ancient times (Ostroverkhova et al. 2017; Kaskinova et al. 2022). Beekeeping as a staple of agriculture in the region is thought to have emerged when external bee colonies were introduced into the area. According to available information (Ilyasov and Kwon 2021), bees were first brought to the Ust-Kamenogorsk region in 1786 and then spread across the Altai Mountains and Siberia. The bees currently inhabiting these regions predominantly belong to M and C lineages (Ostroverkhova et al. 2017; Kaskinova et al. 2022). Subsequently, bees have been imported into Kazakhstan from several locations including Bashkiria, Ukraine, and Orenburg (Baryshnikov et al. 1990). However, according to their phenotype, those bees had likely descended from the European dark bee.
Until now, the impact of indigenous bees on the local beekeeping has been severely underresearched. The discovery of A. m. pomonella in western Tian Shan mountains (Sheppard and Meixner 2003) placed Southeastern Kazakhstan inside the native range of A. mellifera (Alaux et al. 2019; Tihelka et al. 2020). This endemic subspecies is believed to be the result of the crossbreeding between A. mellifera and A. cerana (Liu et al. 2016) and may indicate a step within the westward expansion of the O lineage (Chen et al. 2016).
The main bee subspecies bred in Kazakhstan for commercial use include A. m. sossimai, A. m. carpatica, A. m. mellifera, and A. m. carnica. The primary scope of this research is to reveal the genetic structures of honeybee populations in Kazakhstan, their subspecies-based distribution, and identify a genetically distinct pool for further breeding.
2 Material and methods
2.1 Sampling
Bees (n = 437) were collected from 21 apiaries located predominantly in Southern and Southeastern Kazakhstan, since these regions are characterized by favorable climatic conditions and a flowering plant diversity suitable for beekeeping (Figure 1). Very few bee colonies have been registered in the northern regions, and beekeeping is poorly developed in the west of the country. The subspecies were assigned to colonies based on the beekeepers’ records and documentation, including genetic profiles of the imported bee queens.

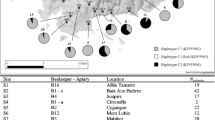
The map of bee sampling locations in Kazakhstan. A Distribution of sampled subspecies and B distribution of detected COI-COII-haplotypes.
The following bee species were identified: A. m. carnica (n = 205), A. m. sossimai (n = 115), A. m. carpatica (n = 64), A. m. caucasica (n = 32), A. m. ligustica (n = 5), and A. m. mellifera (n = 1). Half of the apiaries housed more than one bee subspecies, and all the subspecies were represented in the Tyulbay apiary. Additionally, there were apiaries consisted of each of the subspecies exclusively (Table I), except for A. m. ligustica, which was only found in the populations with three or more subspecies.
The average age of the apiaries was about 5 years, and some of them had genealogical data available. Most of the apiaries were established by bee queens from European lines of A. mellifera L. The apiaries from the Karakonyz gorge, Zhambyl region, and Ugam gorge in Turkistan were established 50 years ago. The apiary from the village of Tokzhailau in the Almaty region is around 25 years old.
The single sample of A. m. mellifera from Belukha Mountain (Russian Federation) and 15 A. m. carnica bees provided by Prof. Dr. Kaspar Bienefeld from the Institute for Bee Research (Hohen Neuendorf, Germany) and Humboldt University (Berlin, Germany) were used as reference groups in genetic analysis to identify possible hybrids and understand the genetic diversity among local populations.
The sample pool was divided according to geographic location, and within each locality the subspecies of each colony was also considered. A total of 21 populations grouped by geographical location and 36 subspecies-based populations were analyzed (Table I and Figure 1).
2.2 DNA extraction
DNA was isolated from a worker bee of every potential colony following a modified CTAB (cetyltrimethylammonium bromide) method (Porebski et al. 1997). Either thorax or legs from every bee were homogenized in liquid nitrogen with further incubation in 1 ml of CTAB buffer (4% CTAB, 100 mM tris (pH 8.0), 1.4 M NaCl, 20 mM ethylenediaminetetraacetic acid (pH 8.0), and 0.4% β-mercantoethanol added before use. The samples were incubated for one hour at 65 °C with a shaking frequency of 1000 rpm. The separation of the water phase containing nucleic acids from the organic contents was performed by adding the same volume of chloroform to homogenized samples. After centrifugation at 13,000 rpm for 15 min, the water phase was transferred to a new tube containing 500 ml of ice-cold 2-propanol and left at 4 °C overnight. Following the precipitation of genomic DNA over 15 min at 13,000 rpm, the DNA pellets were washed with 75% ethanol. The dried pellets were then dissolved in 100 µl of ultra-pure water. The DNA concentration was assayed by spectrophotometry (Nanodrop One) and diluted to 10 ng/µl for PCR. The DNA stocks were preserved at − 80 °C for long-term storage.
2.3 Mitochondrial DNA analysis
The PCR amplification of the intergenic COI-COII region was performed using the E2 (5′-GGCAGAATAAGTGCATTG-3′) and H2 (5′-CAATATCATTGATGACC-3′) primers, following the published protocol (Garnery et al. 1993). The size of the amplicons was determined by electrophoresis in 1.5% agarose gel. To identify the haplotypes, the PCR products were digested by DraI and evaluated on a 10% polyacrylamide gel. The DraI mtDNA COI-COII (DmCC) test was performed according to the protocol described by Garnery et al. (1993).
To validate results of restriction analysis, we sequenced the COI-COII region. PCR was carried out using Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Scientific, MA, USA) followed by the purification of the PCR products with the GeneJET PCR Purification Kit (Thermo Scientific, MA, USA). The sequencing was conducted on a 3500 Genetic Analyzer (Applied Biosystems, CA, USA) according to the manufacturer’s protocol using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA). The obtained consensus sequences of approximately 500 bp were BLAST searched against the NCBI GenBank and compared with the database containing full length COI-COII regions developed by Alburaki et al. (2023). In the case of matches with the same top score, the choice was made in favor the larger haplogroup, i.e., C2.
2.4 Microsatellite analysis
All honeybee specimens were genotyped by nine microsatellite loci (Franck et al. 2001; Delaney et al. 2009). The first multiplex group included markers A24, A28, A43, A88, and A124 (Franck et al. 2001). The other four markers (A007, A113, Ap55, and Ap43) were amplified in groups of two according to their respective protocols (Delaney et al. 2009; Dunin et al. 2019) and analyzed as a multiplex due to their expected fragment sizes and fluorescent dye, to ensure unobstructed detection in each channel and sequence range (Table S1). Every marker was amplified in 20 µl of a reaction mix containing a 1 × Standard Taq Reaction Buffer (M0273E, New England BioLabs, MA, USA), 0.4 µl of 10 mM dNTP, 0.4 µl of 10 µM forward and reverse primers, and 1U Taq-polymerase (M0273E, New England BioLabs, MA, USA); the mix was adjusted to 20 µl using ddH2O.
Amplification was confirmed by agarose gel electrophoresis (1.5% w/v) visualized using a Gel Doc XR+ Imaging System (Bio-Rad, USA). The PCR products were then purified with the GeneJET PCR Purification Kit (Thermo Fisher Scientific, MA, USA) and diluted to 30 ng/µl. Each multiplex group contained the reaction mix including 1 µl of purified PCR product for each marker, 0.15µl of GeneScan 500 LIZ Size Standard (Applied Biosystems, USA), 8.85 µl of HI-DI Formamide (Applied Biosystems, USA), and the reaction mix was adjusted to 50 µl using ddH2O. After denaturation for 4 min at 95 °C, the mix was subjected to capillary electrophoresis on a 3500 Genetic Analyzer (Applied Biosystems, CA, USA) using the FragAnalysis50_POP7 module.
2.5 Statistical and phylogenetic analysis
The genotypes were determined based on their fluorescence peaks in GeneMapper 6 (Applied Biosystems, CA, USA). The population structure was analyzed using the STRUCTURE 2.3.4 software (Pritchard et al. 2000) with the following settings: admixture model with no linkage, prior use of population information, 50,000 burn in, and 100,000 MCMC iterations. The analysis was run for K from 1 to 10 with ten repeats each. The CLUMPAK web server (Kopelman et al. 2015) was used to visualize the results and determine the optimal K value according to Evanno’s method (Evanno et al. 2005). Phylogenetic analysis was conducted using the “adegenet” (Jombart and Ahmed 2011), “poppr” (Kamvar et al. 2014), and “ape” (Paradis et al. 2004) packages in R4.2.1 (R Core Team 2022). The distance matrix was calculated using Nei’s genetic distance and then used to build a neighbor-joining tree. Mantel test implemented in the R package “ecodist” (Goslee and Urban 2007) was used to estimate the correlation between matrices of genetic and geographic distance between populations. The tree was visualized in the FigTree software (Rambaut et al. 2018). The “hierfstat” R package (Goudet 2005) was used to calculate the general population statistics, including the expected (He) and observed (Ho) heterozygosity, inbreeding coefficient Fis, and pairwise fixation index Fst (Weir and Cockerham 1984). Only populations with at least 10 colonies sampled were considered for any population-related calculations. Confidence intervals for Fis and Fst were calculated by bootstrapping with 1000 data permutations. Principal component analysis (PCA) was performed over individual bee samples and populations using the built-in R function “prcomp”.
3 Results
3.1 Screening of mitochondrial haplotypes
DNA amplification and restriction of the COI-COII intergenic region were successful for the entire sample pool (n = 437). European evolutionary lineages C (98.4%) and M (1.6%) were detected in Kazakhstan (Table I). According to the DmCC test, the colonies belonged to C1 (76.4%), C2x (22.2%), and M17 (1.6%) haplogroups (Figure 1). These haplotypes have also been previously described in several studies (Szalanski et al. 2016; Techer et al. 2017), and correspond to the following band patterns found in Figure 2: C1–Q sequence (420, 64, 47, and 41 bp) (Garnery et al. 1993), C2–Q sequence (420, 64, 47, and 40 bp) (Franck et al. 2001), and M17–PQQ sequence (422, 142, 131, 66, and 65 bp) (Rortais et al. 2011; Chávez-Galarza et al. 2017).

Haplotype differentiation by DraI mtDNA COI-COII test. Haplotypes C2, C1, and M17 identified among samples of populations Alakol (A group samples), Kyzylorda-car (G10, G17), Aktas-car (SPK21), Ust-car (243), and A. m. mellifera from the Belukha Mountain (242), respectively. M—Ultra low-range DNA ladder (Invitrogen).
Sequencing of the PCR products allowed us to identify haplotypes more accurately (Table I). A considerable portion of sequences (n = 80) showed great similarity to accessions MT745901-MT745908 from Saudi Arabia without a specific haplotype mentioned in the GenBank, so those were evaluated based on the previously performed RFLP analysis.
Excluding the larger haplogroups C1, C2, and M17, 16 unique haplotypes were identified. Twelve of the studied populations included mixed colonies of both C1 and C2x haplotypes, and only 17 colonies from the Kyzylorda population featured the C1 haplotype as an exclusive mitotype. It was established 32 years ago by the bee queen of Peshetz line 33–00033, and breeding within it has since continued without the addition of new lineages or haplotypes. The M17 haplotype was identified in the sample from the Belukha Mountain and in 60% of the Ushbulak colonies previously classified by us as A. m. sossimai and four as A. m. carnica, indicating an A. m. mellifera queen progenitor. Indeed, Ushbulak apiary was established by a native queen, and thus included colonies of M and C lineages. Most of the populations studied have been formed by the continuous introduction of new queens, whether of the same or different subspecies or races.
The most diverse population in terms of unique genotypes were A. m. sossimai bees from Balambay, which was characterized by the presence of C2j, C2e (C3), C2c, C1, C2jc, and C2d (C2) haplotypes. The most diverse apiary in terms of number of different subspecies, Tyulbay, was characterized by three colonies of C1a haplotype and 17 colonies of C2x haplotypes: C2e (C3) and C2ja. Some authors (Chávez-Galarza et al. 2017) consider C2e haplotype (Muñoz et al. 2009) to be similar to C3 described by (Perrier et al. 2003), while suggesting C2d ought to be renamed C2 due to its high frequency.
3.2 Genetic variability of microsatellite loci
The genetic diversity of 36 bee populations distributed on the territory of Kazakhstan was analyzed using nine informative SSR markers. SSR genotyping data is available in Table S1. All SSR markers were polymorphic, with the number of alleles varying from 5 in the marker A24 to 35 in the marker AP55 (Table II). A total of 158 alleles were detected across the nine loci among all samples, with an average of 17.56 alleles per marker. Fifteen analyzed populations with 10 or more samples displayed higher diversity of alleles than the control group of A. m. carnica from Germany (total 42 alleles, with average 4.67 alleles per marker) (Table III). All calculated genetic parameters of these populations are shown in Table S2. The maximum number of alleles across all loci was identified in the populations of the Kyrgyzai-sos, or the A. m. sossimai colonies from the Kyrgyzai apiary, (69 alleles, 7.67 alleles per marker) and Ugam-cau, or the A. m. caucasica colonies from the Ugam apiary (68 alleles, 7.56 alleles per marker). The former also had nine private alleles detected, which was the highest number across all populations; however, the private alleles were present only occasionally in all populations. Across 14 local populations, the lowest number of detected alleles was observed in the populations of Kokpek-carp (46 alleles, 5.11 alleles per marker) and Shelek-sos (48 alleles, 5.33 alleles per marker). The markers with the highest overall number of detected alleles were AP55 (35 alleles), AP43 (32 alleles), and A124 (25 alleles); the lowest number of alleles was detected in markers A24 (5 alleles) and A28 (7 alleles).
The overall observed heterozygosity (Ho) of the markers ranged from 0.2892 (A28) to 0.9099 (A124). The highest Ho among all the A. m. carnica, was observed in the Alakol-car population for markers A124 (1.0) and AP43 (1.0), and among the A. m. caucasica subspecies, the highest Ho was in the Ugam-cau population for marker A124 (1.0). Marker A28 was represented in Kokpek-carp with the lowest value of Ho (0.0303). The Ho values were slightly deviating from the He values for most markers and individual tested populations; the notable exception was the population Ugam-cau with markers A24, A28, A43, and AP43 having lower Ho. For the markers and populations with the excess of heterozygotes (Table S2, negative values of He–Ho), negative values of the inbreeding coefficient Fis also were observed. Across the populations, the highest average Fis value was in Ugam-cau population (0.1877, within a confidence interval of 0.0197 > Fis > 0.3156; most other populations had positive Fis values below 0.1 or negative values, with confidence intervals including zero, indicating that the Fis values had not significantly differed from zero. The Fis of the total sample was 0.027; however, it was not significant as it lay outside the confidence interval 0.0501 > Fis > 0.1029.
3.3 Genetic relationships among populations
Considering the genetic diversity among the geographically divided populations, the maximum genetic distance (0.5162) was found between Karakonyz-sos and the control group of A. m. carnica from Germany, while the minimum value (0.0442) was identified between the Shelek-sos and Kokpek-carp, according to Nei’s formula (Table S3). A genetic distance matrix for 14 populations with 10 or more samples (excluding the German control group) was compared with the matrix of geographical distances using the Mantel test (Table S3); the test was performed on the whole set of populations with different subspecies, as well as separately for the populations of A. m. carnica (6 populations) and A. m. sossimai (5 populations). In all cases, the Mantel test produced no significant correlations at the confidence level p = 0.05: the corresponding p-values for the two-sided hypothesis (“Mantel’s correlation coefficient r less or greater than zero”) were 0.137, 0.426, and 0.636, respectively.
A phylogenetic tree was constructed using the neighbor-joining method based on Nei’s genetic distance of individuals. The tree confirmed the presence of seven groups and separate individual samples belonging to different subspecies and geographical populations (Figure 3). The samples—U1808 Ugam-cau, U1802 Ugam-sos, and U1803 Ugam-cau—formed the clade with the largest genetic distances to other samples (Figure S2). The largest stable sub-clade included most of A. m. carnica samples from population Akkainar-car, along with a single control sample and a sample of A. m. caucasica from the population Balambay-cau within the clade V. The control samples of A. m. carnica from Germany (shown black) were distributed across various clades; however, most of them were present in a sub-clade within clade III, along with other A. m. carnica samples. In general, this subspecies more tended to form substructures within clades, whereas other subspecies were more dispersed; however, such substructures have reflected the known population structure only to a limited extent.

The phylogenetic tree representing the genetic relationship among Apis mellifera L. samples that was determined based on Nei’s genetic distance.
A principal component analysis of individual samples (Figure 4A) has not revealed significant structure; all subspecies present in the data have formed overlapping groups of points; although the points corresponding to A. m. carpatica (shown yellow) and A. m. caucasica (green) were separated by PC1, their positions were not distinguished from A. m. carnica (red) and A. m. sossimai (purple). A. m. mellifera and A. m. ligustica were represented by only a few samples, thus their genetic structure in respect to other subspecies has remained unclear. The PCA of 15 populations (Figure 4C) was in partial agreement with the neighbor-joining tree for these populations (Figure 4B). All populations of A. m. sossimai, except Shelek-sos were combined into a close cluster along with the population Ugam-sos. Population Shelek-sos was closer to A. m. carnica, along with two populations of A. m. carpatica. Populations Akkainar-car and Shymkent-car were close to other A. m. carnica populations in the tree, however, deviated on the principal component plot.

Principal component analysis (PCA) of individual bee samples and population. A PCA plot of individual samples; B neighbor joining tree of 15 bee populations, based on Nei’s distances; and C PCA plot of 15 bee populations.
The fixation index (FST) values between populations ranged from 0.0118 (Tyulbay-sos/Ugam-cau) to 0.2558 (Karakonyz-sos/Kokpek-car) (Table IV). Comparing the local population with the control group (Germany), the values ranged from 0.0498 for Tokzhailau-car to 0.1728 for Karakonyz-sos. Across all compared pairs of populations, only the partial correspondence between the higher Fst values and the assignment of populations to different subspecies was observed, e.g., A. m. sossimai colonies of Kyrgyzai-sos and Shelek-sos had values 0.1558 and 0.0415, correspondingly, relative to the Kokpek-carp population of A. m. carpatica.
3.4 Cluster analysis
The diversity of populations showed a high level of heterogeneity according to Bayesian cluster analysis. The cluster analysis confirmed the presence of two clusters (max ΔK) (Figure 5). The first cluster comprised five populations, namely the C-group, Kokpek-carp, Akkainar-car, Kargaly-sos, and Shelek-sos. The last two populations were represented by A. m. sossimai and were located close to one another. The second cluster was represented by six populations: Karakonyz (A. m. caucasica and A. m. sossimai), Karakonyz aboriginal, Tyulbay (A. m. caucasica, A. m. carnica and A. m. ligustica), Ugam (A. m. caucasica and A. m. sossimai), Kyrgyzai-sos, Alakol-cau, and A. m. mellifera. The third cluster comprised three populations that showed a background similar to the second cluster, but with a low degree of admixture: Kogaly (A. m. carnica and A. m. ligustica), Alakol-sos, and Balambay. The rest showed strong resemblance to both clusters.

Population structure and genetic differentiation of Apis mellifera L. based on microsatellite markers.
The subspecies could not be differentiated based on the clustering because the same subtypes showed different genetic patterns. However, the geographical locations of the populations had a significant influence on the formation of the clusters (Figure 1 and Table IV). The exceptions were the populations from Alakol; A. m. caucasica and A. m. sossimai were assigned to the second cluster while A. m. carnica formed a mix of two clusters. Considering K = 5 (ΔK = 0.336), only four clusters and eight populations with a single dominant pattern were identified. Nevertheless, K = 5 revealed new genetic patterns for the Kokpek-carp, Shymkent-car, and Kyrgyzai-sos populations. The A. m. carpatica specimens of Kokpek-carp and A. m. sossimai of Shelek-sos differed from the first cluster A. m. carnica of K = 2. The same trend was observed for the A. m. sossimai colonies from Kargaly.
Additionally, one single cluster was represented by the A. m. carnica individuals from the Shymkent-car population. The A. m. carnica colonies of the Akkainar-car population and the A. m. sossimai colonies of Kyrgyzai-sos belonged to the same cluster as the samples from the C-group. However, the queens of Akkainar-car and Kyrgyzai-sos originated from different subspecies and countries, Germany and Ukraine, respectively. The bee colonies of these populations were formed by 47 pure lines of A. m. carpatica for Kokpek-carp and pure lines of A. m. sossimai for Kyrgyzai-sos, originated from Ukraine.
4 Discussion
The importance of increasing the diversity of honeybees in Kazakhstan by the introduction of various subspecies is related with changes of beekeeping conditions, such as the replacement of natural flora with crops, forest reduction, and climate changes; an increase in bee diversity could potentially help overcome the possible negative effects of such factors. On the other hand, this leads to eroding genetic structures of subspecies in populations by occasional and/or systematic hybridization events, and so makes breeding and genotype management more challenging.
The goal of this study was to take a first glance at the genetic background of beekeeping in Kazakhstan, focusing in particular on commercial apiaries, which originated from bees imported throughout the last century. A. m. caucasica was first imported in 1912, while A. m. ligustica and A. m. carnica were introduced into Eastern and Southern Kazakhstan in 1950 (Baryshnikov et al. 1990). A. m. sossimai originated in Ukraine (Engel 1999) and has been broadly distributed in Kazakh apiaries as well. A stock of A. m. mellifera, which has been present since the eighteenth century, has since then formed wild colonies in several regions of the country. Unregulated breeding of the unmanaged A. m. mellifera with the imported subspecies is the possible cause of the mixed genetic nature of honeybee populations in Kazakhstan, and may account for the genetic traces of the M evolutionary lineage. Similarly, the apiaries of pure A. m. mellifera located in the mountains were also affected by random cross-breeding, leading to a decreased in bee productivity and their susceptibility to nosematosis along with other diseases.
Despite amounting to more than 50% of the sample pool, A. m. carnica is second in haplotype diversity in comparison to the colonies classified as A. m. sossimai, or Ukrainian bees. These were characterized by 14 unique haplotypes belonging to all evolutionary lineages present in Kazakhstan. This subspecies was proposed by Engel (1999), and was later suggested to be an ecotype of A. m. macedonica based on mtDNA analysis (Ilyasov et al. 2011), and is still sometimes considered to be a separate species (Ilyasov et al. 2020). As a result, it has not been studied in-depth as either a subspecies or an ecotype, and there are still doubts about its evolutionary lineage. In this study, A. m. sossimai colonies were predominantly characterized by C2x haplotypes. Additionally, the close clustering of Shelek-sos colonies with A. m. carpatica from Kokpek goes back to the morphological analysis done by Ruttner (Ruttner 1988, 1992), who considered A. m. carpatica as another ecotype of A. m. macedonica in the southern part of Romania. However, mtDNA analysis argues the opposite point of view (Coroian et al. 2014; Syromyatnikov et al. 2018).
Overall, C lineage was by far the most dominant across the observed populations, with C2 haplogroup accounting for 9 of the 16 unique haplotypes. The two most-common specific haplotypes found within the populations, C2c (21.3%) and C1a (11.4%), were also widely distributed across their native region (Northern Balkans and Italy, respectively) and are connected to each other by one mutational change (Muñoz and De la Rúa 2021; Oleksa et al. 2021). Notably, the most prominent European C lineage haplotype C2d (C2) was found only in seven of the studied colonies. This distinction suggests that the bees of this haplotype were not imported at the same rate as the rest of the C lineage. These patterns were present in other countries with cases of import such as USA, where C2d remains widely distributed across the country, albeit in low quantity, and served as an evolutionary baseline for the novel haplotypes (Alburaki et al. 2023).
Among the five colonies classified as A. m. ligustica, only one belonged to the C1a haplotype, which characterizes this subspecies (Pinto et al. 2014). Italian honey bee breeders widely export A. m. ligustica queens inside and outside Italy, and most come from Emilia-Romagna, where the C1a haplotype is very common. This can be attributed either to misclassification or uncontrolled breeding between various colonies inside one apiary. Colonies classified as A. m. ligustica were found only in apiaries with diverse subspecies compositions, which hinted at replacement by other dominant subspecies such as A. m. carnica, in accordance with either natural or human selection. A. m. caucasica, which was previously considered to be a part of the O lineage based on morphometry (Kandemir et al. 2011), has been confirmed as a member of the C lineage based on mtDNA analysis (Ilyasov et al. 2019). The results of this study support this thesis further, with 96.8% of colonies belonging to one of the C2x haplotypes.
A DraI COI-COII restriction test, used here for the identification of haplotypes, is a classical tool for the classification and conservation of genetic identity of honeybee populations (Rortais et al. 2011); the sequencing of COI-COII mtDNA region allows more precise haplotype identification (Ajao et al. 2021). However, because of the specific maternal inheritance of mtDNA, COI-COII haplotype analysis has its limitations and should be supplemented with data on nuclear genetics.
The admixture of local honeybee populations has been observed more apparently using the data on SSR genotyping. Unlike the mitochondrial DNA, the nuclear DNA markers are not limited by the maternal lineage and reflect both genetic components of occurring mating events. The genotyping data analyzed using the different methods (PCA, neighbor-joining, STRUCTURE algorithm) have demonstrated the lack of differentiation between a priori defined groups: subspecies and populations. As can be seen from Figure 4, there is a limited impact of geographical location on the differentiation of sampled populations, which could possibly overcome the genetic variation; e.g., populations of four subspecies from Tyulbay (16–19) demonstrated similar patterns of genetic structure. However, the spatial factor has had only limited impact, as the Mantel test has not supported the hypothesis about the correlation between genetic and geographic distances. Principal component analysis, neighbor-joining distance tree, and pairwise fixation indices Fst also indicated the lack of strong patterns of differentiation with respect to subspecies and populations. Although we have named seven clades in the distance-based tree (Figure 3), this grouping was merely arbitrary, as no biological background was identified behind these clusters; the composition of these clades has not reflected known geographic data and/or assumed subspecies memberships. The reasons for such weak genetic structure possibly lay in free genetic flow between colonies of different subspecies and populations. The maximum recorded mating distance for the honeybees was 15 km (Peer 1957; Jensen et al. 2005b). As no studied populations have been located closer to each other than 37.9 km (distance between populations Shelek-sos and Kokpek-carp), the factor of spontaneous hybridization could take place only in populations consisting of multiple subspecies. Hence, the genetic admixture could possibly be explained by undocumented exchanges of bee lineages or other beekeeping practices. Nevertheless, the impact of free mating between bees of different subspecies also should not be neglected: for instance, four subspecies from Tyulbay, as mentioned above, became similar to each other, despite their recorded history. The nearby apiaries, which have not been investigated in this research, could also be a source for outbreeding. As was suggested previously, honeybees are a rare case where human management plays as a factor of increasing rates of admixture and genetic diversity (Harpur et al. 2012). Even the pure, well maintained, and protected breeding populations may be not free of admixture and introgression between subspecies (Jensen et al. 2005a; Soland-Reckeweg et al. 2009).
We cannot rule out the possibility of introgression of wild honeybee subtypes; although the available data are not sufficient, this hypothesis should be tested in further studies. Cross-breeding with A. m. mellifera may take place due to the proximity of the abandoned apiaries colonized by A. m. mellifera to commercial ones (Baryshnikov et al. 1990). As we were able to retrieve only a single specimen of A. m. mellifera from the slope of Belukha Mountain (Russia), the populations could not be compared with A. m. mellifera, and its introgression in the genetic pools of local apiaries could not be evaluated. Moreover, the genetic purity of the dark forest bee specimen could also not be confirmed. Another possible source of admixture is the endemic wild bee subspecies from the regions of the Tian Shan Mountains, A. m. pomonella (Sheppard and Meixner 2003), where most of the studied specimens were collected. However, the absence of available genetic material of A. m. pomonella leaves this hypothesis speculative, until the representative sample of this subspecies could be found and analyzed.
The present study had its limitations related with the incomplete availability of the honeybee genetic material from the studied region; however, it has highlighted some problems of beekeeping and honeybee breeding in the country. To date, no reference populations of any subspecies or breeds have been established in Kazakhstan; most apiaries are private, and therefore, it is almost impossible to determine the exact origin of admixed bee populations. Other problems include (a) absence of established standards and guidelines for classification and control of bee subspecies and/or breeds, (b) uncontrolled breeding on experimental stations and apiaries, (c) extensive undocumented import of different subspecies and/or breeds, (d) abandonment of apiaries without support from the government, (e) lack of genetic profiling for queens and records on breeding. Without addressing these problems, including on the level of the government, the beekeeping and honeybee breeding in Kazakhstan will have low economic efficiency and limited potential for further development. The present work is the first research focused on investigating the genetic structure of bee populations distributed in the largest apiaries of the country. These results would be helpful for planning and performing future investigation and would help lay a basis for genetics-based honeybee breeding in Kazakhstan.
5 Conclusions
According to our findings, honeybee populations in Kazakhstan can be characterized by high level of genetic variability due to the highly admixed genetic pool. Although some local patterns of genetic diversity could be revealed, no strict boundaries between geographic (populations) and genetic groups (subspecies) were revealed. Such promiscuous genetic composition of honeybee populations in Kazakhstan has been assumed to be the result of multiple uncontrolled cross-breeding events; however, the exact sources and mechanisms of admixture should be studied further.
References
Ajao AM, Nneji LM, Adeola AC, Oladipo SO, Ayoola AO, Wang Y-Y, Adeniyi AV, Olademeji YU (2021) Genetic diversity and population structure of the native Western African honeybee (Apis mellifera adansonii Latreille, 1804) in Nigeria based on mitochondrial COI sequences. Zool Anz 293:17–25. https://doi.org/10.1016/j.jcz.2021.05.007
Alaux C, Le Conte Y, Decourtye A (2019) Pitting wild bees against managed honey bees in their native range, a losing strategy for the conservation of honey bee biodiversity. Frontiers in ecology and evolution 7
Alburaki M, Madella S, Lopez J, Bouga M, Chen Y, vanEngelsdorp D (2023) Honey bee populations of the USA display restrictions in their mtDNA haplotype diversity. Frontiers in genetics 13
Alburaki M, Moulin S, Legout H, Alburaki A, Garnery L (2011) Mitochondrial structure of Eastern honeybee populations from Syria, Lebanon and Iraq. Apidologie 42:628–641. https://doi.org/10.1007/s13592-011-0062-4
Baryshnikov S, Rib R, Bashmakov A (1990) The book of beekeeper [Kniga pchelovoda], 1st edn. Kainar, Alma-Ata
Chávez-Galarza J, Garnery L, Henriques D, Neves CJ, Loucif-Ayad W, Jonhston JS, Pinto MA (2017) Mitochondrial DNA variation of Apis mellifera iberiensis: further insights from a large-scale study using sequence data of the tRNAleu-cox2 intergenic region. Apidologie 48:533–544. https://doi.org/10.1007/s13592-017-0498-2
Chen C, Liu Z, Pan Q, Chen X, Wang H, Guo H, Liu S, Lu H, Tian S, Li R, Shi W (2016) Genomic analyses reveal demographic history and temperate adaptation of the newly discovered honey bee subspecies Apis mellifera sinisxinyuan n. ssp. Mol Biol Evol 33:1337–1348. https://doi.org/10.1093/molbev/msw017
Coroian CO, Muñoz I, Schlüns EA, Paniti-Teleky OR, Erler S, Furdui EM, Mărghitaş LA, Dezmirean DS, Schlüns H, de la Rúa P, Moritz RFA (2014) Climate rather than geography separates two European honeybee subspecies. Mol Ecol 23:2353–2361. https://doi.org/10.1111/mec.12731
Cridland JM, Tsutsui ND, Ramírez SR (2017) The complex demographic history and evolutionary origin of the western honey bee, Apis Mellifera. Genome Biol Evol 9:457–472. https://doi.org/10.1093/gbe/evx009
Delaney DA, Meixner MD, Schiff NM, Sheppard WS (2009) Genetic characterization of commercial honey bee (Hymenoptera: Apidae) populations in the United States by using mitochondrial and microsatellite markers. Ann Entomol Soc Am 102:666–673. https://doi.org/10.1603/008.102.0411
Dogantzis KA, Tiwari T, Conflitti IM, Dey A, Patch HM, Muli EM, Garnery L, Whitfield CW, Stolle E, Alqarni AS, Allsopp MH, Zayed A (2021) Thrice out of Asia and the adaptive radiation of the western honey bee. Science advances 7:eabj2151. https://doi.org/10.1126/sciadv.abj2151
Dogantzis KA, Zayed A (2019) Recent advances in population and quantitative genomics of honey bees. Current Opinion in Insect Science 31:93–98. https://doi.org/10.1016/j.cois.2018.11.010
Dunin I, Novikov A, Kalashnikova L, Kalashnikov A, Gladyr E (2019) Guidelines: the standard methods of genetic identification, determining the validity of origin and appraisal of honey bee colonies (translation) [Metodicheskie rekomendacii: standartnye metody geneticheskoj identifikacii, opredeleniya dostovernosti proiskhozhdeniya i bonitirovki pchelinyh semej]. VNIIplem, Lesnye Polyany
Engel MS (1999) The taxonomy of recent and fossil honey bees (Hymenoptera: Apidae; Apis). J Hymenopt Res 8:165–196
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Franck P, Garnery L, Loiseau A, Oldroyd BP, Hepburn HR, Solignac M, Cornuet J-M (2001) Genetic diversity of the honeybee in Africa: microsatellite and mitochondrial data. Heredity 86:420–430. https://doi.org/10.1046/j.1365-2540.2001.00842.x
Garnery L, Solignac M, Celebrano G, Cornuet J-M (1993) A simple test using restricted PCR-amplified mitochondrial DNA to study the genetic structure of Apis mellifera L. Experientia 49:1016–1021. https://doi.org/10.1007/BF02125651
Goslee SC, Urban DL (2007) The ecodist Package for dissimilarity-based analysis of ecological data. J Stat Soft 22:1–19. https://doi.org/10.18637/jss.v022.i07
Goudet J (2005) hierfstat, A package for r to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Harpur BA, Minaei S, Kent CF, Zayed A (2012) Management increases genetic diversity of honey bees via admixture. Mol Ecol 21:4414–4421. https://doi.org/10.1111/j.1365-294X.2012.05614.x
Ilyasov R, Nikolenko A, Tuktarov V, Goto K, Takahashi J-I, Kwon HW (2019) Comparative analysis of mitochondrial genomes of the honey bee subspecies A. m. caucasica and A. m. carpathica and refinement of their evolutionary lineages. J Apic Res 58:567–579. https://doi.org/10.1080/00218839.2019.1622320
Ilyasov RA, Kutuev I, Petuknov A (2011) Phylogenetic relationships of dark European honeybees Apis mellifera L. from the Russian Ural and west European populations. Journal of Apicultural Science 55
Ilyasov RA, Kwon HW (2021) Phylogenetics of Bees, 1st edn. CRC Press, Boca Raton, FL
Ilyasov RA, Lee M, Takahashi J, Kwon HW, Nikolenko AG (2020) A revision of subspecies structure of western honey bee Apis mellifera. Saudi Journal of Biological Sciences 27:3615–3621. https://doi.org/10.1016/j.sjbs.2020.08.001
Jensen AB, Palmer KA, Boomsma JJ, Pedersen BV (2005a) Varying degrees of Apis mellifera ligustica introgression in protected populations of the black honeybee, Apis mellifera mellifera, in northwest Europe. Mol Ecol 14:93–106. https://doi.org/10.1111/j.1365-294X.2004.02399.x
Jensen AB, Palmer KA, Chaline N, Raine NE, Tofilski A, Martin SJ, Pedersen BV, Boomsma JJ, Ratnieks FLW (2005b) Quantifying honey bee mating range and isolation in semi-isolated valleysby DNA microsatellite paternity analysis. Conserv Genet 6:527–537. https://doi.org/10.1007/s10592-005-9007-7
Ji Y (2021) The geographical origin, refugia, and diversification of honey bees (Apis spp.) based on biogeography and niche modeling. Apidologie 52:367–377. https://doi.org/10.1007/s13592-020-00826-6
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. https://doi.org/10.1093/bioinformatics/btr521
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kandemir İ, Özkan A, Fuchs S (2011) Reevaluation of honeybee (Apis mellifera) microtaxonomy: a geometric morphometric approach. Apidologie 42:618–627. https://doi.org/10.1007/s13592-011-0063-3
Kaskinova MD, Gaifullina LR, Kinzikeev AK, Gladkih AN, Sokolyanskaya MP, Saltykova ES (2022) Genetic structure of the Apis mellifera L. population from Altai Krai. Russ J Genet 58:880–883. https://doi.org/10.1134/S1022795422070092
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Liu F, Shi T, Huang S, Yu L, Bi S (2016) Genetic structure of Mount Huang honey bee (Apis cerana) populations: evidence from microsatellite polymorphism. Hereditas 153:8. https://doi.org/10.1186/s41065-016-0010-4
Muñoz I, Dall’Olio R, Lodesani M, De la Rúa P, (2009) Population genetic structure of coastal Croatian honeybees (Apis mellifera carnica). Apidologie 40:617–626. https://doi.org/10.1051/apido/2009041
Muñoz I, De la Rúa P (2021) Wide genetic diversity in old world honey bees threaten by introgression. Apidologie 52:200–217. https://doi.org/10.1007/s13592-020-00810-0
Oleksa A, Kusza S, Tofilski A (2021) Mitochondrial DNA suggests the introduction of honeybees of African ancestry to East-Central Europe. InSects 12:410. https://doi.org/10.3390/insects12050410
Ostroverkhova NV, Kucher AN, Konusova OL, Kireeva TN, Sharakhov IV (2017) Genetic diversity of honeybees in different geographical regions of Siberia. International Journal of Environmental Studies
Paradis E, Claude J, Strimmer K (2004) APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. https://doi.org/10.1093/bioinformatics/btg412
Peer DF (1957) Further studies on the mating range of the honey bee, Apis mellifera L. Can Entomol 89:108–110. https://doi.org/10.4039/Ent89108-3
Perrier C, Strange J, Langella O, Sheppard W, Garnery L (2003) Diversité génétique, introgressions mitochondriales et nucléar dans une population d’abeilles des Landes de Gascogne. Les Actes Du BRG 4:79–100
Pinto MA, Henriques D, Chávez-Galarza J, Kryger P, Garnery L, van der Zee R, Dahle B, Soland-Reckeweg G, de la Rúa P, Dall’ Olio R, Carreck NL, Johnston JS, (2014) Genetic integrity of the dark European honey bee (Apis mellifera mellifera) from protected populations: a genome-wide assessment using SNPs and mtDNA sequence data. J Apic Res 53:269–278. https://doi.org/10.3896/IBRA.1.53.2.08
Porebski S, Bailey LG, Baum BR (1997) Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15:8–15. https://doi.org/10.1007/BF02772108
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
R Core Team (2022) R: A language and environment for statistical computing
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032
Rortais A, Arnold G, Alburaki M, Legout H, Garnery L (2011) Review of the DraICOI-COII test for the conservation of the black honeybee (Apis mellifera mellifera). Conservation Genet Resour 3:383–391. https://doi.org/10.1007/s12686-010-9351-x
Ruttner F (1988) Biogeography and Taxonomy of Honeybees. Springer, Berlin, Heidelberg
Ruttner F (1992) Naturgeschichte der Honigbienen. Ehrenwirth, Munich
Sheppard WS, Meixner MD (2003) Apis mellifera pomonella, a new honey bee subspecies from Central Asia. Apidologie 34:367–375. https://doi.org/10.1051/apido:2003037
Soland-Reckeweg G, Heckel G, Neumann P, Fluri P, Excoffier L (2009) Gene flow in admixed populations and implications for the conservation of the Western honeybee, Apis mellifera. J Insect Conserv 13:317–328. https://doi.org/10.1007/s10841-008-9175-0
Syromyatnikov MY, Borodachev AV, Kokina AV, Popov VN (2018) A molecular method for the identification of honey bee subspecies used by beekeepers in Russia. InSects 9:10. https://doi.org/10.3390/insects9010010
Szalanski AL, Tripodi AD, Trammel CE, Downey D (2016) Mitochondrial DNA genetic diversity of honey bees, Apis mellifera, in Hawaii. Apidologie 47:679–687. https://doi.org/10.1007/s13592-015-0416-4
Techer MA, Clémencet J, Simiand C, Preeaduth S, Azali HA, Reynaud B, Hélène D (2017) Large-scale mitochondrial DNA analysis of native honey bee Apis mellifera populations reveals a new African subgroup private to the South West Indian Ocean islands. BMC Genet 18:53. https://doi.org/10.1186/s12863-017-0520-8
Tihelka E, Cai C, Pisani D, Donoghue PCJ (2020) Mitochondrial genomes illuminate the evolutionary history of the Western honey bee (Apis mellifera). Sci Rep 10:14515. https://doi.org/10.1038/s41598-020-71393-0
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. https://doi.org/10.2307/2408641
Whitfield CW, Behura SK, Berlocher SH, Clark AG, Johnston JS, Sheppard WS, Smith DR, Suarez AV, Weaver D, Tsutsui ND (2006) Thrice Out of Africa: ancient and recent expansions of the honey beE, Apis mellifera. Science 314:642–645. https://doi.org/10.1126/science.1132772
Acknowledgements
We sincerely thank Prof. Dr. Kaspar Bienefeld from the Institute for Bee Research Hohen Neuendorf and the Humboldt University of Berlin for providing fifteen samples of A. m. carnica used as control group in the current research.
Funding
This research was funded by the Ministry of Agriculture of the Republic of Kazakhstan, grant number BR10764957 “Development of technologies for effective management of the Breeding process in beekeeping” and BR10765038 “Development of methodology and implementation of scientifically based system of certification and inspection of seed potatoes and planting material of fruit crops in the Republic of Kazakhstan”.
Author information
Authors and Affiliations
Contributions
DG conceptualized the study. KT isolated the DNA. AP and MK analyzed the statistics and wrote the manuscript. AT and VK performed experiments. MKh prepared the maps. OK, AM, GM, KT, and UN collected samples.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material/data availability
The data is available in supplementary materials.
Code availability
Not applicable.
Additional information
Manuscript editor: Klaus Hartfelder.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gritsenko, D., Temirbayeva, K., Taskuzhina, A. et al. First evaluation of genetic diversity among honeybee populations in Kazakhstan. Apidologie 54, 61 (2023). https://doi.org/10.1007/s13592-023-01034-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13592-023-01034-8