
Abstract
Osteosarcoma (OSA), the malignant bone tumor, predominantly affecting children and adolescents, threatens the life and life quality of the patients. An increasing number of studies have indicated the role of long non-coding RNA (lncRNA) dysregulation in cancer biology. Herein, the study was aimed to explore the role of FGD5 antisense RNA 1 (FGD5-AS1), a lncRNA, in OSA. Expression levels of FGD5-AS1, miR-506-3p and RAB3D mRNA were quantified utilizing qRT-PCR. The expression of RAB3D protein was examined employing Western blot. A series of functional experiments including CCK-8 assay, BrdU assay, wound healing assay, Transwell assay were performed for studying the effects of FGD5-AS1 on the malignancy of OSA cell lines 143B and HOS. The binding site between miR-506-3p and FGD5-AS1 was identified and validated by luciferase reporter assay and RNA immunoprecipitation assay. It was demonstrated that the expression of FGD5-AS1 was up-regulated in OSA tissues and cell lines, and its high expression is associated with higher Enneking stage and poorer histological differentiation. Gain-of-function and loss-of-function studies suggested that FGD5-AS1 facilitated OSA cells proliferation and migration. The promoting effects of FGD5-AS1 overexpression on OSA cell proliferation and migration could be counteracted by miR-506-3p. Moreover, FGD5-AS1 competitively adsorbed miR-506-3p to repress its expression so as to up-regulate the expression of RAB3D. These results indicate that FGD5-AS1 is capable of expediting OSA cell proliferation and migration via sponging miR-506-3p to up-regulate RAB3D.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteosarcoma (OSA), the primary malignancy of bone, is a common tumor among children and adolescents [1]. Unfortunately, approximately 10–20% of the cases are with distant metastasis when they are diagnosed [2]. The current 5-year survival rate of OSA remains lower than 60%; even though some of the patients can survive, a large proportion of the patients received amputation, which seriously reduces the life quality [3]. Therefore, clarifying the mechanism of OSA progression for exploring novel therapy strategies is an imperative demand.
Long non-coding RNA (lncRNA), consisting of more than 200 nucleotides, has attracted fast-growing attention in the field of cancer research over the past few years [4]. An increasing number of lncRNAs are reported to participate in the regulation of biological processes of cancer cells, including cell proliferation, metastasis, and apoptosis [5,6,7,8]. In the progression of OSA, lncRNAs are also important players. A lot of dysregulated lncRNAs, including MALAT1, SNHG3, and CBR3-AS1, have been reported to be associated with the development of OSA [9,10,11]. However, the expression, function, and underlying mechanisms of lncRNA FGD5 antisense RNA 1 (FGD5-AS1) remain unexplained in OSA.
Herein, the expression, clinical significance, biological functions, and underlying mechanisms of FGD5-AS1 in OSA were investigated. It was demonstrated that FGD5-AS1 was significantly up-regulated in OSA tissues and cells, and its high expression facilitate the malignant biological phenotypes of OSA cells. Additionally, it was verified as a molecular sponge of miR-506-3p, and up-regulates oncogene RAB3D.
Materials and methods
Patients and specimens
Tissue samples from 50 patients with OSA (8–28 years old; 37 males, 13 females; from 2012 May to 2019 March) were collected. OSA tissues and their matched non-cancerous cartilage tissues were procured, then immediately cryopreserved in liquid nitrogen. This study was approved by the Ethics Committee of People’s Hospital of Rizhao and informed consents were obtained from all patients. All patients’ sections were reviewed to confirm the diagnosis and to classify the tumor according to Enneking staging system. The clinical and pathological features of the patients are shown in supplementary table 1.
Cell culture
Human OSA cell lines (143B, HOS, U2OS, and MG63) and normal osteoblast cell lines (hFOB 1.19) were purchased from China Center for Type Culture Collection (Wuhan, China). All cells were cultivated in RPMI-1640 medium (Invitrogen, Waltham, MA, USA) containing 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA), placed in an incubator at 37 °C with 5% CO2 and saturated humidity.
Cell transfection
FGD5-AS1 overexpression vector (pcDNA3.1-FGD5-AS1), control plasmid (pcDNA3.1), FGD5-AS1 siRNA (si-FGD5-AS1#1, si-FGD5-AS1#2), scramble siRNA (si-NC), miR-506-3p mimics (miR-506-3p), miR-506-3p inhibitors (miR-506-3p in), and negative control mimics (miR-NC) were designed and provided by GenePharma (Shanghai, China). Transfection was conducted using Lipofectamine®2000 (Invitrogen, Waltham, MA, USA) according to the protocol provided by the manufacturer. The sequences were listed as below: si-FGD5-AS1#1, 5′-CAUUUGUAAUAGUGUUCAAUA-3′; si-FGD5-AS1#2: 5′-GCGUAGUUACAAUGAUUUAAA-3′; si-NC: 5′-UUCUCCGAACGUGUCACGUTT-3′; miR-506-3p mimics: 5′-UAAGGCACCCUUCUGAGUAGA-3′; miR-506-3p in: 5′-UCUACUCAGAAGGGUGCCUUA-3′; miR-NC, 5′-UCACAACCUCCUAGAAAGAGUAGA-3’.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA in tissues and cells was obtained with a TRIzol kit (Invitrogen, Carlsbad, CA). PrimeScript RT regent kit (TakaraBio, Tokyo, Japan) was employed to synthesize cDNA. SYBR Premix Ex Taq II (ABI, Foster City, CA, USA) was used in DNA amplification. GAPDH served as the internal reference of FGD5-AS1 and RAB3D, and U6 served as the internal reference of miR-506-3p. Relative expressions of the genes were analyzed by 2−ΔΔCT formula. The primers designed were detailed in Table 1. For nuclei/cytoplasm separation assay, the nuclear/cytoplasmic RNA was isolated from 143B and HOS cells using a PARIS™ Kit following the protocol and subjected to qRT-PCR analysis, respectively, to detect the RNA enrichment in the nuclei and cytoplasma.
Cell counting kit-8 (CCK-8) assay
OSA cells in the logarithmic growth phase were trypsinized with 0.25% trypsin for preparation of single-cell suspension and inoculated into 96-well plates (2 × 103/well), and cultured for 24, 48, 72, 96 h, respectively. At each time point, 10 μL of CCK-8 reagent (Dojindo, Kumamoto, Japan) was added to each well and then incubated for additional 2 h. After that, the absorbance value of each well at 450 nm wavelength was detected by a microplate reader.
BrdU assay
Transfected cells (5 × 103/well) were inoculated in 96-well plates, cultivated at 37 °C for 24 h, and labeled with BrdU reagent (BD Pharmingen, San Diego, CA, USA). Cellular DNA was denatured, then BrdU primary antibody was added and the cells were incubated at room temperature for 2 h. After that, the cells were added with secondary antibody, and incubated for 2 h at ambient temperature, and re-stained nucleus with DAPI staining solution, followed by observation under a fluorescence microscope. Ten non-overlapping fields of vision were randomly selected. The number of BrdU positive cells was counted and averaged.
Wound healing assay
After the cells reached about 90% confluence, the tip of the 200 μL sterilized pipette was used to make a scratch on the cells in the middle of the dish vertically. Subsequently, the cells were washed with serum-free medium for 3 times. Then the intervals between the cells were observed and photographed. After that, the cells were cultured in serum-free medium for 24 h, the intervals between the cells were observed and photographed again. The change of width of the intervals was used to evaluate the migration ability of OSA cells.
Transwell assay
Cell migration was also measured utilizing a double chamber Transwell system with a pore size of 8 mm (Biosharp, Hefei, China). 200 μL of prepared cell suspension (1 × 105 cells /mL, suspended in serum-free medium) was dripped into the upper chamber, and 500 μL of RPMI-1640 medium containing 10% FBS was supplemented to the lower compartment. The cells were cultured for 24 h. Then the cells on the below surface of the membrane were fixed with methanol and stained with 0.1% crystal violet solution. Then the stained cells were counted, by which the migration ability of tumor cells was evaluated.
Western blot
RIPA lysis buffer (Beyotime, Shanghai, China) was applied for protein extraction, and a BCA kit (Beyotime, Shanghai, China) was employed for protein quantification. The protein samples were dissolved by 10% SDS–polyacrylamide gel electrophoresis and next the separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). The unspecific antigens were blocked at room temperature for 2 h in 5% defatted milk, and then the membrane was incubated with anti-RAB3D primary antibody (Abcam, Cambridge, UK) at 4 °C overnight. Thereafter, the membrane was incubated with HRP-labeled secondary antibody for additional 2 h at room temperature (Abcam, Cambridge, UK). Ultimately, development of protein bands was performed employing an enhanced chemiluminescence detection system (Bio-Rad, Hercules, CA, USA).
Luciferase reporter assay
The binding site between FGD-AS1 and miR-506-3p was available by online bioinformatics analysis tool StarBase (http://starbase.sysu.edu.cn/). The FGD5-AS1 sequence containing the putative binding site of miR-506-3p was inserted into psi-CHECK2 reporter vector (Promega, Madison, WI, USA), generating luciferase reporter vector FGD5-AS1-wild-type (FGD5-AS1-WT), and correspondingly, luciferase reporter vector FGD5-AS1 mutant (FGD5-AS1-MUT) was constructed. HEK-293 cells were co-transfected with FGD5-AS1-WT or FGD5-AS1-MUT and miR-506-3p or miR-NC. 24 h later, the luciferase activity of the cells was examined with a dual-luciferase reporter system (Promega, Madison, WI, USA).
RNA immunoprecipitation (RIP) assay
EZ-Magna RIP™ RNA-binding protein immunoprecipitation kit (Millipore, Billerica, MA, USA) was applied for detecting the direct binding relationship between FGD5-AS1 and miR-506-3p. Cells were lysed using RIP buffer, and then the cell lysate was incubated with anti-Argonaute2 (Ago2) antibody or normal immunoglobulin G (IgG) at 4 °C overnight. Then the immunoprecipitate was incubated with proteinase K. Subsequently, total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Finally, the relative enrichment of FGD-AS1 and miR-506-3p was determined by qRT-PCR analysis.
Lung metastasis model in vivo
Our animal experiments were approved by the Animal Research Ethics Committee of People's Hospital of Rizhao. 40 six-week-old male BALB/c nude mice were purchased from the Slack Jingda Experimental Animal Co., Ltd. (Changsha, China). 143B and HOS cells transfected with pcDNA3.1-FGD5-AS1 or FGD5-AS1 siRNA were injected into nude mice via caudal vein (1 × 107 cells per mouse). Three weeks later, the mice were killed, and the lungs were collected. Following that, HE staining was performed, and the metastatic nodules were evaluated under a microscope. Severe metastasis: the metastases ≥ 50% of observer's field of view; moderate metastasis: the metastases < 50% of observer's field of view.
Statistical analysis
Each experiment was performed with three replicates, and repeated for a minimum of three times, and the data were presented as the “mean ± standard deviation”. All statistical analyses were carried out utilizing SPSS 20.0 (SPSS Inc., Chicago, IL, USA). The difference between two groups was compared by Student’s t test. P < 0.05 indicated statistical significance.
Results
FGD5-AS1 shows up-regulated expression in OSA and it was associated with clinical features
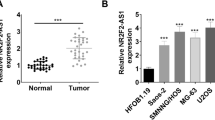
FGD5-AS1 expression was detected in 50 paired OSA samples and adjacent normal tissues by qRT-PCR, the findings of which revealed it was markedly up-regulated in malignant tissues compared with peritumoral tissues (Fig. 1a, P < 0.001). Additionally, FGD5-AS1 expression was markedly up-regulated in OSA cell lines compared with hFOB1.19 cells (Fig. 1b, P < 0.01). The associations between FGD5-AS1 expression in OSA samples and clinicopathological features were further analyzed. It showed that FGD5-AS1 up-regulation was associated with higher Enneking stage, poorer tissues differentiation (Fig. 1c, d, P < 0.05), suggesting that FGD5-AS1 overexpression might be involved in the progression of OSA.

Expression of FGD5-AS1 in OSA tissues and cell lines. a Expression level of FGD5-AS1 in OSA tissues (n = 50) and adjacent normal tissues (n = 50) was analyzed via qRT-PCR. b Expression level of FGD5-AS1 in OSA cell lines and the normal human osteoblastic cell line, hFOB, was determined by qRT-PCR. c, d qRT-PCR was used to analyze the expression of FGD5-AS1 in patients with different clinical pathological features. All of the experiments were performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001
FGD5-AS1 overexpression expedites proliferation, migration, and metastasis of OSA cells
Next, gain-of-function and loss-of-function cells models were established with 143B and HOS cells, respectively (Fig. 2a, P < 0.001). CCK-8 assay and BrdU assay showed that, compared with the control group, ectogenic expression of FGD5-AS1 notably potentiated the viability of 143B cells, whereas FGD5-AS1 silencing markedly reduced the viability of HOS cells (Fig. 2b, c, P < 0.01). Additionally, wound healing assay and Transwell assay manifested that FGD5-AS1 overexpression facilitated the migration of 143B cells (Fig. 3a, b, P < 0.01). Conversely, FGD5-AS1 silencing suppressed the migration of HOS cells (Fig. 3a, b, P < 0.01). Additionally, in vivo experiments showed that, FGD5-AS1 overexpression promoted the lung metastasis of OSA cells, while the depletion of FGD5-AS1 repressed the formation of metastatic nodules in the lung (Fig. 4, P < 0.05). Collectively, these findings indicated that FGD5-AS1 overexpression expedited the progression of OSA.

Effects of FGD5-AS1 on the proliferation of osteosarcoma cells. a qRT-PCR was conducted to detect the expression of FGD5-AS1 in 143B and HOS cells after transfection. b CCK-8 assay was employed to assess cell viability of 143B and HOS cells after transfection. c BrdU assay was employed to assess cell proliferation of 143B and HOS cells after transfection. All of the experiments were performed in triplicate. **P < 0.01, ***P < 0.001

Effects of FGD5-AS1 on the migration of osteosarcoma cells. a, b Wound healing assay and cell migration assay were conducted to detect cell migration of the transfected 143B and HOS cells. All of the experiments were performed in triplicate. **P < 0.01, ***P < 0.001

FGD5-AS1 promoted the metastasis of OSA cells in vivo. 143B were transfected with FGD5-AS1 overexpression plasmids or empty vector, and HOS cells were transfected with si-FGD5-AS1 or si-NC, then lung metastasis models were established with nude mice (n = 10 in each group), and HE staining was used to evaluate the severity of the lung metastasis
FGD5-AS1 adsorbs miR-506-3p and constrains its expression
We further extracted RNA from 143B and HOS cells by nucleoplasm separation and found that FGD5-AS1 was mainly located in the cytoplasm of OSA cells, suggesting that it might participate in OSA progression mainly through post-transcriptional regulation (Fig. 5a). Reportedly, lncRNA can function as a competing endogenous RNA (ceRNA) or a molecular sponge, interact with miRNAs and participate in regulating physiological and pathological processes. Accordingly, StarBase database (http://starbase.sysu.edu.cn/index.php) was employed in our research for predicting the potential miRNA targets for FGD5-AS1, and the results indicated that FGD5-AS1 possessed a putative binding site for miR-506-3p (Fig. 5b). Dual-luciferase reporter assay suggested that the luciferase activity of FGD5-AS1-WT reporter vector was partly inhibited by miR-506-3p; after that binding site was mutated by site-directed mutagenesis, the inhibitory function of miR-506-3p on the luciferase activity of reporter vector was abolished (Fig. 5c, P < 0.001). What is more, RIP experiment suggested that FGD5-AS1 was preferentially enriched in Ago2-containing immunoprecipitates (Fig. 5d, P < 0.001). Additionally, miR-506-3p expression was remarkably down-regulated in cancer tissues and OSA cell lines as opposed to normal tissues and cells (Fig. 5e, f, P < 0.05). FGD5-AS1 overexpression repressed miR-506-3p expression in 143B cells, whereas FGD5-AS1 silencing augmented miR-506-3p expression in HOS cells (Fig. 5g, P < 0.01). Furthermore, Pearson’s correlation analysis showed that miR-506-3p expression was negatively correlated with FGD5-AS1 expression in OSA samples (Fig. 5h, P < 0.001). These data supported that FGD5-AS1 could serve as a miRNA sponge, regulating miR-506-3p.

FGD5-AS1 could bind with miR-506-3p and suppressed its expression in OSA. a The enrichment of FGD5-AS1 in cytoplasm and nuclei of 143B and HOS cell lines was analyzed. b Bioinformatics analysis showed that miR-506-3p was predicted to be a target miRNA of FGD5-AS1. c Dual-luciferase reporter assays showed that miR-506-3p could negatively regulate the luciferase activity of FGD5-AS1-WT, rather than FGD5-AS1-MUT. d The binding relationship between FGD5-AS1 and miR-506-3p was validated by RIP assay. qRT-PCR was used to detect the FGD5-AS1 enrichment in the immunoprecipitation in miR-506-3p-overexpressing OSA cells. e qRT-PCR was conducted to detect the expression of miR-506-3p in OSA tissues and adjacent tissues. f Expression of miR-506-3p in OSA cell lines and the normal osteoblastic cell line were analyzed via qRT-PCR. g qRT-PCR showed that FGD5-AS1 could negatively regulate miR-506-3p expression in OSA cells. h Pearson’s correlation analysis revealed there was a negative correlation between miR-506-3p expression and FGD5-AS1 expression in OSA tissues. All of the experiments were performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001
FGD5-AS1 facilitates OSA cell proliferation and migration by targeting miR-506-3p/RAB3D axis
As shown, transfection of miR-506-3p mimics induced a decrease in expression of RAB3D, while FGD5-AS1 overexpression restored RAB3D expression at mRNA and protein levels in HOS cells (Fig. 6a, b, P < 0.01). OSA cell viability was inhibited by transfection of miR-506-3p mimics, and the effects of miR-506-3p were counteracted by FGD5-AS1 overexpression (Fig. 6c, d, P < 0.05). MiR-506-3p repressed the migration of HOS cells, and this effect was reversed by elevated FGD5-AS1 expression (Fig. 5e, f, P < 0.05). As expected, after miR-506-3p was inhibited, the expression of RAB3D was up-regulated, and co-transfection of FGD5-AS1 siRNA counteracted this in 143B cells (Fig. 5a, b, P < 0.01). MiR-506-3p inhibition promoted the malignancy of 143B cells, while co-transfection of FGD5-AS1 siRNA rescued these phenomena (Fig. 6c–f, P < 0.05). Additionally, the up-regulation of RAB3D induced by FGD5-AS1 was reversed by si-RAB3D (Fig. 7a, b). The effect of FGD5-AS1 overexpression on the proliferation and migration of 143B cells was weakened by si-RAB3D (Fig. 7c–f). Collectively, by targeting miR-506-3p, FGD5-AS1 indirectly up-regulated RAB3D’s expression, thereupon facilitating OSA progression.

FGD5-AS1 promoted OSA cell proliferation and migration by targeting miR-506-3p. a, b qRT-PCR and Western blot were conducted, respectively, to analyze the expression of RAB3D mRNA and protein levels in miR-NC group, miR-506-3p mimics group and miR-506-3p m.imics + FGD5-AS1 group. c, d CCK-8 assay and BrdU assay were employed to assess cell proliferation of the transfected 143B and HOS cells in miR-NC group, miR-506-3p mimics group and miR-506-3p mimics + FGD5-AS1 group. e, f Wound healing assay and migration assay were employed to assess cell migration of the transfected 143B and HOS cells in miR-NC group, miR-506-3p mimics group and miR-506-3p mimics + FGD5-AS1 group. All of the experiments were performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001

FGD5-AS1 facilitates OSA cell proliferation and migration depending on RAB3D. a, b qRT-PCR and Western blot were conducted, respectively, to analyze the expression of RAB3D mRNA and protein levels in 143B cells in vector group, FGD5-AS1 group, and FGD5-AS1 + si-RAB3D group. c, d CCK-8 assay and BrdU assay were employed to assess cell proliferation of the transfected 143B cells in vector group, FGD5-AS1 group, and FGD5-AS1 + si-RAB3D group. e, f Wound healing assay and migration assay were employed to assess cell migration of the transfected 143B in vector group, FGD5-AS1 group, and FGD5-AS1 + si-RAB3D group. All of the experiments were performed in triplicate. **P < 0.01, ***P < 0.001
Discussion
The non-protein-coding portion of human genome was previously considered as junk DNA. Amongst the non-protein-coding transcripts, lncRNAs have attracted increasing attention in recent years. Nowdays, lncRNA has been proved to regulate gene expression at transcriptional or post-transcriptional level by multiple mechanisms, which is associated with their capability of interacting with DNA, RNA, or protein [12,13,14]. At the transcriptional level, lncRNAs may participate in modulation of chromatin structures by recruiting chromatin-modifying enzymes, resulting in expression or repression of genes; during post-transcriptional regulation, lncRNAs regulate mRNA stability, splicing, modifications and so on [6, 15]. The relationship between the abnormal expression of lncRNA and cancer biology has been gradually recognized [16,17,18]. Previous studies have reported that lncRNAs regulate cell proliferation, invasion, and chemotherapeutic drug resistance of OSA cells [19,20,21,22,23]. For example, SPRY4‑IT1 accelerates proliferation, cell cycle progression, migration, and invasion of OSA cells via regulating ZEB1 and ZEB2 expression trough sponging of miR-101 [20]. MALAT1 promotes OSA cell viability, invasion, and migration [21]. What’s more, ASB16-AS1 expression was associated with tumor size, TNM stage, and distant metastasis; the overall survival rate of OSA patients presenting high ASB16-AS1 expression was lower [22]. FGD5-AS1 has been reported to exert a pivotal role in human cancers. For instance, inhibition of FGD5-AS1 blocks cancer cell proliferation, migration, invasion, and induces cell apoptosis in colorectal cancer [24]. Highly expressed FGD5-AS1 facilitates non-small cell lung cancer cell proliferation by sponging miR-107 and up-regulating FGFRL1 [25]. In the current study, it was revealed that FGD5-AS1 was up-regulated in OSA tissues and cell lines, and its high expression was associated with unfavorable pathological characteristics of the patients. Furthermore, FGD5-AS1 overexpression expedited proliferation and migration of OSA cells, while FGD5-AS1 knockdown reduced the malignancy of cancer cells, indicating that FGD5-AS1 exerted a promoting role in regulating the oncogenic properties of OSA cells and participated in OSA progression.
MicroRNAs (miRNAs), characterized as small non-coding RNAs that repress translation and stability of mRNA, modulates the expression levels of multiple genes, which are involved in regulating cell proliferation, inflammation, cell cycle progression, stress response, differentiation, apoptosis, and migration. Their dysregulation exert a crucial role in the progression of cancer [26,27,28]. Reportedly, miR-506-3p is down-regulated in various cancers. Overexpression of miR-506-3p restrains prostate cancer progression by targeting GALNT4 in vitro [29]. MiR-506-3p also suppresses cell proliferation, induces cell cycle arrest and apoptosis in retinoblastoma by directly targeting NEK6 [30]. A previous study identifies miR-506-3p serves as a tumor suppressor in OSA and its down-regulation contributes to tumor cell proliferation and metastasis by regulating RAB3D- and CDK4-mediated signaling [31]. Another study reports that miR-506-3p directly targets SPHK1 to inhibit the epithelial-mesenchymal transition (EMT) of OSA cells [32]. ceRNA networks link the function of protein-coding mRNAs with that of non-coding RNAs such as microRNA, long non-coding RNA, pseudogenic RNA and circular RNA [33,34,35,36]. In the present study, miR-506-3p, consistent with previous published studies, was down-regulated in OSA tissues and cells. FGD5-AS1 was demonstrated to contain a binding site for the seed sequence of miR-506-3p and FGD5-AS1 could negatively regulate the expression of miR-506-3p. MiR-506-3p expression exhibited a negative correlation with FGD5-AS1 in OSA tissues. Based on these findings, it was concluded that FGD5-AS1 served as a ceRNA for miR-506-3p to regulate its expression in OSA cells.
RAB3D, a member of ras-related GTP-binding protein Rab family, is identified to be a regulator of intracellular vesicle transport during exocytosis. Some studies indicate that RAB3D is in association with the transcytotic pathway [37, 38]. The expression of RAB3D shows dysregulation in cancers. RAB3D activates Akt/GSK3β/Snail pathway and induces EMT process in colorectal cancer cells [39]. Besides, RAB3D functions as an oncogene in esophageal squamous cell carcinoma and knockdown of RAB3D impeded cancer cell proliferation and invasion via disrupting PI3K/Akt signaling [37]. Additionally, RAB3D is highly expressed in OSA tissues and OSA cell lines, and it facilitates the growth and metastasis of OSA cells [31, 40]. RAB3D has been reported to be a target of miR-506-3p in OSA cells [31]. In the present study, consistently, qRT-PCR and Western blot indicated that miR-506-3p negatively regulated expression of RAB3D mRNA and protein, while FGD5-AS1 counteracted the inhibitory effect of miR-506-3p on RAB3D mRNA and protein expression, substantiating that the function of FGD5-AS1/ miR-506-3p axis in OSA is partly mediated by RAB3D.
To conclude, FGD5-AS1/miR-506-3p/RAB3D axis regulates the malignant biological behaviors of OSA cells. These findings will improve the understanding of the molecular mechanism involved in OSA progression and provide novel targets for the molecular treatment of OSA.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Kager L, Tamamyan G, Bielack S. Novel insights and therapeutic interventions for pediatric osteosarcoma. Future Oncol. 2017;13:357–68.
Liu YJ, Li W, Chang F, et al. MicroRNA-505 is downregulated in human osteosarcoma and regulates cell proliferation, migration and invasion. Oncol Rep. 2018;39:491–500.
Simpson S, Dunning MD, de Brot S, et al. Comparative review of human and canine osteosarcoma: morphology, epidemiology, prognosis, treatment and genetics. Acta Vet Scand. 2017;59:71.
Wang J, Su Z, Lu S, et al. LncRNA HOXA-AS2 and its molecular mechanisms in human cancer. ClinChimActa. 2018;485:229–33.
Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77:3965–81.
Peng WX, Koirala P, Mo YY. LncRNA-mediated regulation of cell signaling in cancer. Oncogene. 2017;36:5661–7.
Wei GH, Wang X. lncRNA MEG3 inhibit proliferation and metastasis of gastric cancer via p53 signaling pathway. Eur Rev Med PharmacolSci. 2017;21:3850–6.
Liu Y, Yang Y, Li L, et al. LncRNA SNHG1 enhances cell proliferation, migration, and invasion in cervical cancer. Biochem Cell Biol. 2018;96:38–43.
Ren D, Zheng H, Fei S, et al. MALAT1 induces osteosarcoma progression by targeting miR-206/CDK9 axis. J Cell Physiol. 2018;234:950–7.
Zheng S, Jiang F, Ge D, et al. LncRNA SNHG3/miRNA-151a-3p/RAB22A axis regulates invasion and migration of osteosarcoma. Biomed Pharmacother. 2019;112:108695.
Zhang Y, Meng W, Cui H. LncRNA CBR3-AS1 predicts unfavorable prognosis and promotes tumorigenesis in osteosarcoma. Biomed Pharmacother. 2018;102:169–74.
Jiang R, Tang J, Chen Y, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. 2017;8:15129.
Liu XH, Sun M, Nie FQ, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92.
Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407.
Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18:206.
Sanchez Calle A, Kawamura Y, Yamamoto Y, et al. Emerging roles of long non-coding RNA in cancer. CancerSci. 2018;109:2093–100.
Loewen G, Jayawickramarajah J, Zhuo Y, et al. Functions of lncRNA HOTAIR in lung cancer. J HematolOncol. 2014;7:90.
Kumar MM, Goyal R. LncRNA as a therapeutic target for angiogenesis. Curr Top Med Chem. 2017;17:1750–7.
Yang Z, Li X, Yang Y, et al. Long noncoding RNAs in the progression, metastasis, and prognosis of osteosarcoma. Cell Death Dis. 2016;7:e2389.
Yao H, Hou G, Wang QY, et al. LncRNA SPRY4-IT1 promotes progression of osteosarcoma by regulating ZEB1 and ZEB2 expression through sponging of miR-101 activity. Int J Oncol. 2020;56:85–100.
Duan G, Zhang C, Xu C, et al. Knockdown of MALAT1 inhibits osteosarcoma progression via regulating the miR-34a/cyclin D1 axis. Int J Oncol. 2019;54:17–28.
Yin R, Liu J, Zhao D, et al. Long non-coding RNA ASB16-AS1 functions as a miR-760 sponge to facilitate the malignant phenotype of osteosarcoma by increasing HDGF expression. Onco Targets Ther. 2020;13:2261–74.
Yan L, Wu X, Yin X, et al. LncRNA CCAT2 promoted osteosarcoma cell proliferation and invasion. J Cell Mol Med. 2018;22:2592–9.
Li D, Jiang X, Zhang X, et al. Long noncoding RNA FGD5-AS1 promotes colorectal cancer cell proliferation, migration, and invasion through upregulating CDCA7 via sponging miR-302e. Vitro Cell Dev BiolAnim. 2019;55:577–85.
Fan Y, Li H, Yu Z, et al. Long non-coding RNA FGD5-AS1 promotes non-small cell lung cancer cell proliferation through sponging hsa-miR-107 to up-regulate FGFRL1. 2020. Biosci Rep. https://doi.org/10.1042/BSR20193309.
Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314.
Chen G, Zhou H. MiRNA-708/CUL4B axis contributes into cell proliferation and apoptosis of osteosarcoma. Eur Rev Med PharmacolSci. 2018;22:5452–9.
Yuan G, Zhao Y, Wu D, et al. miRNA-20a upregulates TAK1 and increases proliferation in osteosarcoma cells. Future Oncol. 2018;14:461–9.
Hu CY, You P, Zhang J, et al. MiR-506-3p acts as a novel tumor suppressor in prostate cancer through targeting GALNT4. Eur Rev Med PharmacolSci. 2019;23:5133–8.
Wu L, Chen Z, Xing Y. MiR-506–3p inhibits cell proliferation, induces cell cycle arrest and apoptosis in retinoblastoma by directly targeting NEK6. Cell BiolInt. 2018. https://doi.org/10.1002/cbin.11041.
Jiashi W, Chuang Q, Zhenjun Z, et al. MicroRNA-506-3p inhibits osteosarcoma cell proliferation and metastasis by suppressing RAB3D expression. Aging. 2018;10:1294–305.
Wang D, Bao F, Teng Y, et al. MicroRNA-506-3p initiates mesenchymal-to-epithelial transition and suppresses autophagy in osteosarcoma cells by directly targeting SPHK1. BiosciBiotechnolBiochem. 2019;83:836–44.
Qi X, Zhang DH, Wu N, et al. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52:710–8.
Wang Y, Zeng X, Wang N, et al. Long noncoding RNA DANCR, working as a competitive endogenous RNA, promotes ROCK1-mediated proliferation and metastasis via decoying of miR-335-5p and miR-1972 in osteosarcoma. Mol Cancer. 2018;17:89.
Cao J, Han X, Qi X, et al. TUG1 promotes osteosarcoma tumorigenesis by upregulating EZH2 expression via miR-144-3p. Int J Oncol. 2017;51:1115–23.
Han N, Zuo L, Chen H, et al. Long non-coding RNA homeobox A11 antisense RNA (HOXA11-AS) promotes retinoblastoma progression via sponging miR-506-3p. Onco Targets Ther. 2019;12:3509–17.
Zhang J, Kong R, Sun L. Silencing of Rab3D suppresses the proliferation and invasion of esophageal squamous cell carcinoma cells. Biomed Pharmacother. 2017;91:402–7.
Millar AL, Pavios NJ, Xu J, et al. Rab3D: a regulator of exocytosis in non-neuronal cells. HistolHistopathol. 2002;17:929–36.
Luo Y, Ye GY, Qin SL, et al. High expression of Rab3D predicts poor prognosis and associates with tumor progression in colorectal cancer. Int J Biochem Cell Biol. 2016;75:53–62.
Cao K, Fang Y, Wang H, et al. ThelncRNA HOXA11-AS regulates Rab3D expression by sponging miR-125a-5p promoting metastasis of osteosarcoma. Cancer Manag Res. 2019;11:4505–18.
Funding
This study was supported by Scientific research support fund for Teachers of Jining Medical College. Project Name: Study on the correlation between mir-202-5p site, XIAP and other related proteins in osteosarcoma (ID: JYFC2018FKJ034).
Author information
Authors and Affiliations
Contributions
BW and CL designed the study. CL and XL conducted most of the experiments and wrote the manuscript. CZ, LW, and JY conducted the experiments and analyzed the data.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
Our study was approved by the Ethics Review Board of People's Hospital of Rizhao (ID: 201703036).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, C., Lin, X., Zhang, C. et al. Long non-coding RNA FGD5-AS1 enhances osteosarcoma cell proliferation and migration by targeting miR-506-3p/RAB3D axis. Human Cell 34, 1255–1265 (2021). https://doi.org/10.1007/s13577-021-00536-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-021-00536-w