
Abstract
Artemisinin a natural sesquiterpenoid isolated from Artemisia annua, is of great interest in the treatment of malaria. In this study important artemisinin and related metabolites were profiled throughout the defined developmental period by utilizing efficient reversed-phase HPLC method with UV detection in two elite varieties CIM-Arogya and Jeevan raksha of Artemisa annua L. Further to gain insight in to the regulation of artemisinin biosynthesis and accumulation during plant ontogeny, expression profiling of eight genes of artemisinin biosynthetic pathway were performed in both the varieties. We found higher expression of all transcripts of artemisinin biosynthetic pathway during plant ontogeny. High level of up regulation of these genes during plant ontogeny was associated with higher level of secondary metabolite content. Relative dynamics of artemisinin metabolites followed an increasing pattern till the month of November. A. annua CIM-Arogya variety had higher accumulation mainly in upper leaves. Interestingly, dihydroartemisinic acid accumulation was at variance with that of artemisinin and arteannuin B, suggesting later months of development preferred equilibrium towards artemisinin accumulation mediated through uncharacterized catalytic transformation. This behavioural dynamics might have some eco-physiological role to play for the plant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Artemisinin is a naturally occurring anti-malarial sesquiterpene lactone endoperoxide which is isolated from Artemisia annua L. belonging to family Asteraceae. The biosynthesis of sesquiterpenoid artemisinin is almost completely elucidated (Fig. 1). There is extensive development in the information regarding biosynthesis, genomics and pathway understanding in A. annua (Towlers and Weathers 2007; Schramek et al. 2010; Singh et al. 1990; Sangwan et al. 1993, 1998, 2010). The isopentenyl pyrophosphate for artemisinin originates from both cytosolic and plastidic isoprenogenesis. Briefly, farnesyl diphosphate synthase catalyzes the synthesis of farnesyl diphosphate from two units of IPP and one unit of dimethyl allyl pyrophosphate (DMAPP), as for all sequiterpenoids and triterpenoids. Catalytic conversion of farnesyl diphosphate into amorpha-4,11-diene through amorpha diene synthase activity constitutes the committed step for biosynthesis of artemisinin and congener metabolites of the pathway artemisinic acid and arteannuin B. In metabolic sequence, amorpha-4,11-diene is sequentially oxidized to generate artemisinic acid involving a single cytochrome P450 enzyme, CYP71AV1, (Teoh et al. 2006). Recently a double bond reductase (DBR2; Zhang et al. 2008) and an aldehyde dehydrogenase (Teoh et al. 2006) involved in the biosynthesis were also isolated. These enzymes are involved in conversion of artemisinic aldehyde to its dihydro form and then to dihydroartemisinic acid, respectively. Aldh1 also convert artemisinic aldehyde to artemisinic acid, an activity ascribed to CYP (Zhang et al. 2008). Dihydro derivative may be converted to artemisinin as shown in Fig. 1 (Brown 2010). Dihydroartemisinic acid is late precursor of artemisinin (Brown 2010) while artemisinic acid is converted to arteannuin B. These conversions may involve non enzymatic or photooxidation/singlet oxygen reaction steps as well (Brown 2010; Sangwan et al. 1993).

Artemisinin biosynthetic pathway
Because of the chemical properties of artemisinin, mainly its low extinction coefficient, various detectors other than UV detection have been employed for quantification, including high-pressure liquid chromatography–evaporative light scattering detection (HPLC–ELSD) (Peng et al. 2006; Liu et al. 2007; Yadav et al. 2013), thin layer chromatography (Marchand et al. 2008) with chemical visualisation, liquid chromatography–mass spectroscopy (LC-MS) (Van Nieuwerburgh et al. 2006) and nuclear magnetic resonance (NMR) (Castilho et al. 2008). Quantification with gas chromatography (GC) is also known (Ferreira and Gonzalez 2008; Peng et al. 2006), but due to the thermal instability of artemisinin, this technique measures the breakdown products of artemisinin and is therefore an indirect technique for making quantitative measurements. Therefore, we utilized a modified HPLC-PDA method that can be used in the efficient quantitative analysis of artemisinin (AN), dihydroartemisinic acid (DHAA) and arteannuin B (AAB) (Yadav et al. 2013). In the present work, the profiling of AN, DHAA and AAB in two elite and released varieties of A. annua (cv. CIM-Arogya and cv. Jeevanraksha) was performed and dynamics of these metabolites with pathway gene expression was studied. Gene-expression profiling of eight-selected target genes, which are involved in artemisinin biosynthesis during developmental stages vis-a-vis metabolites revealed a direct co-relation of gene-to-metabolite link in two cultivar of A. annua throughout developmental period.
Materials and methods
Plant materials
Both the elite varieties of A. annua cv Jeevanraksha and A. annua cv. CIM-Arogya were grown under glasshouse conditions with temperature ranging from 20 to 35 °C, from start of May to end of November under natural light. Plant leaf samples were collected throughout entire developmental period with one month interval for metabolite and gene expression analysis.
Preparation of extract for HPLC
Artemisinin (AN), dihydroartemisinic acid (DHAA) and arteannuin B (AAB) were extracted by using hexane. Extraction was conducted by refluxing 1 g of oven-dried (40–45 °C) leaves of different ages of greenhouse plants in 50 ml hexane for 12 h at 25 °C. Then the n-hexane phase were filtered and evaporated to dryness in a fume hood. The dried extract was reconstituted in 1 ml of HPLC grade methanol and centrifuged at 12,000 g to remove the impurities. The supernatant was filtered through the 0.2 μ filter for HPLC analysis. The analysis was performed on Waters HPLC system equipped with a 515 programmable pump, in-line degasser AV, 717plus autosampler, and 2996 photodiode array detector and reversed phase column (Nova Pack, C18, 4 μm, 150 mm × 3.9 mm). The system was controlled and data analysis was performed with Empower software.
Calibration
The linear detection range for each compound was established by preparing individual standard stock solutions in HPLC grade methanol. Aliquots of these solutions were diluted and analysed to determine the linearity range of the method. Limit of quantification (LOQ) values were estimated from serial dilution and analyzed for each sample. Triplicate 10 μL injections were made for each standard solution to see the reproducibility of the detector response at each concentration level. The peak area of each drug was plotted against the concentration to obtain the calibration graph. The five concentrations of each compound were subjected to regression analysis to calculate calibration equation and correlation coefficients.
HPLC analysis
HPLC analysis was performed as described earlier (Yadav et al. 2013). AN was first derivatized to Q260 for detection and quantification while its precursor’s DHAA and other end product AAB were detected and quantified in intact form without modification. The artemisinin detection and quantification was done by the method of Zhao and Zeng (1985) with modifications. Zhao and Zeng used 55/45 (by volume) methanol/0.01 M phosphate buffer as the mobile phase to determine artemisinin content in crude plant extracts. Artemisinin which lacks any chromophors was first derivatized to Q260 by chemical modification using 0.2 % NaOH and 0.08 M glacial acetic acid. The mobile phase consisted of methanol and water with 0.1 % glacial acetic acid each in ratio of 30:70. Isocratic elution was chosen to achieve maximum separation and sensitivity. Flow rate was 0.8 ml/min. The samples were detected at 260 nm using photodiode array detector. Column condition was same as for artemisinin. The mobile phase consisting of methanol and water with 0.1 % glacial acetic acid each in ratio of 30:70 by isocratic elution was chosen to achieve maximum separation and sensitivity.
Linearity
Ten different concentrations of AN, DHAA, AAB were analyzed and their calibration curve was constructed in the specified concentration range (0.01–0.10 mg/mL). The calibration plots were generated by replicate analysis (n = 3) at all concentration levels and the linear relationship was evaluated using the least square method within Microsoft Excel ®program.
Limit of detection and quantification
The limit of detection (LOD) and limit of quantification (LOQ) were calculated from the calibration curves as kσ/S, where k = 3.3 for LOD, and k = 10 for LOQ, σ is the standard deviation of the Y-intercept of regression line and S is the slope of the calibration curve. Both LOD and LOQ were expressed by the detection quantity (μg) of AN, DHAA, AAB. LOD and LOQ were experimentally verified by six injections of each metabolite at the LOD and LOQ concentrations.
RNA isolation, cDNA synthesis and quantitative real-time PCR analysis
Total RNA was isolated from the tissues by TRIZOL reagent (Invitrogen) according to manufacturer’s instructions. RNA was quantified by NanoDrop ND-1000 (Nanodrop Technologies), and verified for integrity by electrophoresis on a 0.8 % (w/v) agarose gel. A 5 μg aliquot of total RNA was reverse-transcribed into cDNA using the high-capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real time PCR was performed in triplicate on an Applied Biosystem Step One™ Real-Time PCR system using TaqMan primer and probe as described earlier (Yadav et al. 2013). The relative gene expression was determined based on the 2-∆∆CT method. 18 s rRNA was used as an endogenous control gene.
Statistical analysis
Student’s t and one way ANOVA test was conducted for statistical analysis of the data performed. Mean values were compared and standard deviation was indicated except where it was too small to be visible in the graphical representations. Asterisks indicate a significant difference at *P < 0.05.
Results and discussion
Artemisinin absorbs at the lower end of the UV spectrum, between 210 and 220 nm, the extinction coefficient is relatively small and detection at lower wavelengths usually results in poor selectivity (Chen et al. 2002). Although a method was developed for the quantification of AN using HPLC with UV detection at 210–220 nm (Ferreira and Gonzalez 2008), this detection at the low wavelength region has its obvious drawbacks, especially with plant extracts. Zeng et al. (1983) reported that treating artemisinin with sodium hydroxide solution gave a resultant compound, called Q292, having a maximum absorbance at 292 nm. Q292 could be further converted into another compound, called Q260, having a maximum absorbance at 260 nm, by acidifying its solution. Although Q292 has an absorbance maximum at 292 nm with ɛ value of 19400 in alkaline medium (Zeng et al. 1983) it is not very stable so as not to be determined by conventional chromatographic methods. Consequently various HPLC methods with either pre- or post-column derivatization have been developed to convert artemisinin into Q260 owing to its stability around neutral toward slightly acidic pH (Chen et al. 2002). However Q260 (with ɛ value of 11200 in acidic medium) is less sensitive than that of Q292. Therefore Q260 could be used to determine the artemisinin content. In the present work AN was first derivatized to Q260 for detection and quantification while its precursors DHAA and other end product AAB were detected and quantified in intact form without derivatization. DHAA and AAB absorbs at low end of the UV spectrum around 210 nm. Good linearity for AN (r2 = 0.9977), DHAA (r2 = 1), and AAB (r2 = 0.9997) was found between 0.01 mg/ml to 0.1 mg/ml. The LOD was calculated to be 2.06 μg/ mL; the LOQ was calculated to be 6.89 μg/mL for artemisinin, for AAB LOD was 1.6 μg/mL and LOQ was 5.51 μg/mL and for dihydroartemisinic acid LOD was 0.13 μg/mL and LOQ was 0.455 μg/mL.
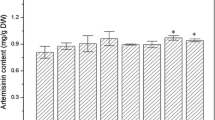
Maes et al. (2011), and Arsenault et al. (2010) also reported the HPLC-ELSD technique for detection and quantification of artemisinic acid, DHAA, and arteannuin B from A. annua samples. Mannan et al. (2010) used LC/MS for AA detection and quantification. The report of artemisinic acid detection and quantification by HPLC–ELSD was also available in literature (Ferreira et al. 1994; Ferreira and Gonzale 2008). In the present study we quantified all the three metabolites in two cultivar of A. annua (cv. CIM-Arogya and cv. Jeevanraksha) during entire plant ontogeny and among different plant tissues such as leaf, flower, shoot and root using efficient protocol (Yadav et al 2013). AN content was higher for cv. CIM-Arogya as compared to cv. Jeevanraksha although the difference is not statistically significant at p < 0.05 level. AN content was obtained at lowest at early vegetative stage (May) and reached its peak at late vegetative state (September) and reproductive stage (October, November) for both cultivars of A. annua (Figs. 1 and 2a). Similarly, AAB content was lowest at early vegetative stage (May) and maximum at late vegetative stage (September) and reproductive stage (October, November) for both cultivars of A. annua (Fig. 2c, Table 1). Several reports are available on the developmental profile of metabolites in different accession of A. annua with the observation that AN concentration peaked just before flowering (Laughlin 1995; Sangwan et al 1999, 2010; Yadav et al. 2013). Nevertheless, observations of some investigation (Ferreira and Janick 1996) suggest it to reach maximum at full flowering. This difference could be attributable to the climatic conditions, ecotype, cultural practices or a combination of all these factors. DHAA is the immediate precursor of artemisinin. The distribution pattern of DHAA during plant ontogeny was different from both AN and AAB. DHAA content in plant was lowest at early vegetative stage (May) and full bloom stage (November) and highest at mid vegetative stage (July). This distribution pattern is attributable for both cultivar of A. annua (Fig. 2b, Table 1). Peng et al. (2006) reported that the AN content of the sixteen seed-generated lines of the cultivar A. annua ranged from 0.2 % to 0.9 % by both GC-FID and HPLC-ELSD. ElSohly et al. (1987) work stated that artemisinin (0.138 %, 0.140 %, and 0.153 %) in A. annua cultivars analyzed by HPLC analysis reached to its maximum amounts prior to flowering period. In an earlier study, artemisinin content in the herb of A. annua was found 0.65 % using RP-HPLC (Qian et al. 2005). Our present study shows that artemisinin percentage varies during plant ontogeny and reached maximum from late vegetative stage (September) to reproductive stage (October, November). The highest artemisinin percentage for A. annua cv. Jeevan Raksha was 1.1 % and for cv. CIM-Arogya was 1.3 % on dry weight basis. Artemisinin distribution estimation was also done in upper leaf (upper 1/3 of plant canopy) and lower leaf (lower 2/3 of plant canopy) and among the plant different tissues (leaf, flower, shoot and root). Upper leaf of plant exhibited around 10–20 % higher artemisinin content compared with lower leaf canopy and the pattern is reflected for both cultivar of A. annua (Fig. 2d and e). The variation of artemisinin concentration down the plant profile varies considerably with origin. Charles et al. (1990), reported about double AN concentration in top third of plant compared from the middle and lower third when sampled in the vegetative phase just before flowering in an accession of A. annua of Chinese origin. Laughlin (1995) reported increase in AN concentration from top to bottom quarters. Ferreira and Janick (1996) reported even distribution of AN in top, middle and lower third at both vegetative and reproductive stage of plant. Plant population density, accession, climatic condition, ecotype and all in combination may be an important factor in analysing above results of variation (Laughlin 1995; Charles et al. 1990; Ferreira et al. 1994; Ferreira and Janick 1996; Bose et al. 2013). In the present study we have taken tissues from each quarter of plant height for artemisinin estimation and found higher AN concentration in leaf tissue from upper 1/3 of plant height compared with remaining 2/3 of plant height. The statement was also validated by real time quantification of transcripts of AN biosynthetic pathway. Interestingly, flower tissues have around 9–18 % lower AN content compared to upper leaf tissues (Table 1). Side shoot branches contained around 0.04 % AN compared with upper leaf, main stem had trace amount of AN while root tissue have detected none (Table 1). Thus the leaf tissues are the main source of AN in plant A. annua and younger leaves representing upper canopy of the plant represent elevated biosynthetic machinery for terpenoid biosynthesis (Singh et al. 1990).

Comparative profiles of artemisinin (a), dihydroartemisinic acid (b), and arteannuin B (c) content (mg g−1 DW) in two cultivars of A annua (cv Jeevanraksha and cv. CIM-Arogya) during plant ontogeny from early vegetative stage (May) to full bloom stage (November). d and e represent the comparative analysis of artemisinin in upper leaf (upper 1/3 of plant) and lower leaf (lower 2/3 of plant) for cultivar CIM-Arogya and Jeevan Raksha respectively. Mean values are compared. Standard deviation is represented as error bar (n = 3). Asterisk indicates the significant difference at level p < 0.05
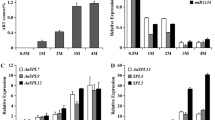
To determine the expression patterns of transcripts of AN biosynthetic pathway to correlate with those of the metabolite analyzed, the gene expression of the selected eight enzymes of metabolism (Table 2) were studied in leaf tissues of A. annua from May to November (Fig. 3a1–a8). Two of the eight enzymes of the MEP pathway, DXS and DXR catalyzed regulatory steps of the pathway (Cordoba et al. 2009). The higher expression of these two genes, as observed by qPCR, during plant ontogeny implies an increase in the metabolic flux through the pathway positively affecting the biogenesis of artemisinin. Recently, it has been shown that AN biosynthesis occurs substantially or wholly through MEP pathway (Schramek et al. 2010). One enzyme of the mevalonate pathway, hydroxymethyl glutaryl co A reductase that has been analyzed for its expression is a rate-limiting enzyme of the mevalonate pathway. The expression of the key pathway gene HMGR was found up-regulated during the plant ontogeny. It has been shown that HMGR expression influences artemisinin formation in A. annua (Ram et al. 2010). Similar to HMGR, DXS AND DXR, the expression of IPP isomerase and FDS were found up-regulated during plant ontogeny. IPP isomerase catalyses the inter conversion of IPP and DMAPP, the key intermediates that are used to produce GPP (precursor of monoterpenes) and FDP, precursor of artemisinin (Yadav et al. 2013). FDS plays a central role in catalyzing FDP synthesis that serves as an anaplerotic metabolite catering to the needs of these diversified pathways. Over expression of FDS in A. annua has been shown to result in an increase in artemisinin production (Ferreira and Janick 1996) indicating that it holds rate control or flux facilitation role in plant. Our data on FDS are also in its close agreement. ADS and CYP71AV1 (ADMO and ADCO) catalyze three steps of AN biosynthesis from FDP and were up-regulated during plant ontogeny and reached its plateau at later stages of plant development. The observed higher expression of ADS and CYP71AV1 involved in the conversion of FDP to AA and DHAA (Brown and Sy 2004) provides expression genomics cue to subdued production of AN and AAB during plant ontogeny. The relative quantification of selected eight genes was also conducted for leaf tissues and flower tissues to correlate the metabolite with transcript dynamics (Fig. 4a and b). All genes indirectly involved in artemisinin biosynthesis (HMGR, DXS, DXR, IPP isomerise and FDS) were found 2–5 fold up-regulated in flower tissue while genes of specific artemisinin biosynthetic (ADS, ADMO and ADCO) pathway were 1–2 fold down regulated. Thus, in conclusion the gene expression data are in correlation with the artemisinin data. Higher expression of HMGR, DXS, DXR, IPP isomerase and FDS in flower tissue indicated higher biosynthesis of monoterpene and sesquiterpene of essential oil, triterpenes and sterols (Sangwan et al. 2010; Yadav et al. 2013). The maximum artemisinin content was obtained at late vegetative stage and reproductive stage for both cultivars. Relatively maximum artemisinin content was higher for cv. CIM-Arogya (1.3 %) than cv. Jeevanraksha (1.1 %). Artemisinin content is higher in upper leaves compared with lower leaves in A. annua suggesting younger and developing leaves to have elevated levels of biosynthetic potential. The levels of the artemisinic metabolites correlated with the pathway gene expression in both the elite varieties.

Relative quantification profiles of eight genes (HMGR: a1, DXS:a2, DXR:a3, IPP isomerase:a4, FDS:a5, ADS:a6, ADMO:a7 and ADCO:a8) of artemisinin biosynthetic pathway during plant ontogeny (May to November) for two cultivar of A. annua (CIM-Arogya and Jeevan Raksha)

Relative expressions of eight genes of artemisinin biosynthetic pathway in A. annua. a expression of the genes in leaf and flower tissues at flowering stage; b expression of the genes in upper leaf (upper 1/3 of plant) and lower leaf (lower 2/3 of plant). Standard deviation is represented as error bar (n = 3). Asterisk indicates the significant difference at level p < 0.05
Abbreviations
- AAB:
-
Arteannuin B
- AN:
-
Artemisinin
- DHAA:
-
Dihydroartemisinic acid
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
References
Arsenault PR, Vail D, Wobbe KK, Erickson K, Weathers PJ (2010) Reproductive development modulates gene expression and metabolite levels with possible feedback inhibition of artemisinin in Artemisia annua. Plant Physiol 154:958–968
Bose SK, Yadav RK, Mishra S, Sangwan RS, Singh AK, Mishra B, Srivastava AK, Sangwan NS (2013) Effect of gibberellic acid and calliterpenone on plant growth attributes, trichomes, essential oil biosynthesis and pathway gene expression in differential manner in Mentha arvensis L. Plant Physiol Biochem 66C:150–158
Brown GD (2010) The biosynthesis of artemisinin (Qinghaosu) and the phytochemistry of Artemisia annua L (Qinghasu). Molecules 15:7603–7698
Brown GD, Sy LK (2004) In vivo transformations of dihydroartemisinic acid in Artemisia annua plants. Tetrahedron 60:1139–1159
Castilho PC, Gouveia SC, Rodrigues AI (2008) Quantification of artemisinin in Artemisia annua extracts by 1 H-NMR. Phytochem Anal 19:329–333
Charles DJ, Simon JE, Wood KV, Heinstein P (1990) Germplasm variation in artemisinin content of Artemisia annua using an alternative method of artemisinin analysis from crude plant extract. J Nat Prod 53:157–160
Chen LH, Wang KT, Pu QS, Chen XG, Hu ZD (2002) Online conversion and determination of artemisinin using a flow-injection capillary electrophoresis system. Electrophoresis 23:2865–2871
Cordoba E, Salmi M, León P (2009) Unravelling the regulatory mechanisms that modulate the MEP pathway in higher plants. J Exp Bot 60:2933–2943
ElSohly HN, Croom EM, ElSohly MA (1987) Analysis of the antimalarial sesquiterpene artemisinin in Artemisia annua by high-performance liquid chromatography (HPLC) with post column derivatization and ultraviolet detection. Pharma Res 4:258–260
Ferreira JFS, Gonzalez JM (2008) Analysis of underivatizatized artemisinin and related sesquiterpene lactones by high-performance liquid chromatography with ultraviolet detection. Phytochem Anal 20:91–97
Ferreira JFS, Janick J (1996) Distribution of artemisinin in Artemisia annua. In: Janick J (ed) Progress in new crops. ASHS Press, Arlington, pp 578–579
Ferreira JFS, Charles DJ, Wood K, Janick J, Simon JE (1994) A comparison of gas chromatography and high performance liquid chromatography for artemisinin analyses. Phytochem Anal 5:116–120
Laughlin JC (1995) The influence of distribution of anti-malarial constituents in Artemisia annua L. on time and method of harvest. Acta Hort 390:67–73
Liu CZ, Zhou HY, Zhao Y (2007) An effective method for fast determination of artemisinin in Artemisia annua L. by high performance liquid chromatography with evaporative light scattering detection. Anal Chim Acta 581:298–302
Maes L, VanNieuwerburgh FCW, Zhang Y, Reed DW, Pollier J, VandeCasteele SRF, Inze D, Covello PS, Deforce DLD, Gossens A (2011) Dissection of the phytohormonal regulation of trichome formation and biosynthesis of the antimalarial compound artemisinin in Artemisia annua plants. New Phytol 189:176–189
Mannan A, Liu C, Arsenault PR, Towler MJ, Vail DR, Lorence A, Weathers PJ (2010) DMSO triggers the generation of ROS leading to an increase in artemisinin and dihydroartemisinic acid in Artemisia annua shoot cultures. Plant Cell Rep 29:143–152
Marchand E, Atemnkeng MA, Vanermen S, PlaizierVercammen J (2008) Development and validation of a simple thin layer chromatographic method for the analysis of artemisinin in Artemisia annua L plant extracts. Biomed Chrom 22:454–459
Peng CA, Ferreira JFS, Wood AJ (2006) Direct analysis of artemisinin from Artemisia annua L. using high-performance liquid chromatography with evaporative light scattering detector, and gas chromatography with flame ionization detector. J Chroma A 1133:254–258
Qian GP, Yang YW, Ren QL (2005) Determination of artemisinin in Artemisia annua L. by reversed phase HPLC. J Liq Chroma Rel Tech 28:705–712
Ram M, Khan MA, Jha P, Khan S, Kiran U, Ahmad MM, Javed S, Abdin MZ (2010) HMG-CoA reductase limits artemisinin biosynthesis and accumulation in Artemisia annua L. plants. Acta Physiol Planta 32:859–866
Sangwan RS, Agarwal K, Luthra R, Thakur RS, Sangwan NS (1993) Biotransformation of arteannuic acid into arteannuin B and artemisinin in Artemisia annua. Phytochemistry 34:1301–1302
Sangwan NS, Sangwan RS, Kumar S (1998) Isolation of genomic DNA from antimalarial plant Artemisia annua. Plant Mole Biol Rep 16:365–372
Sangwan RS, Sangwan NS, Jain DC, Kumar S, Ranade SA (1999) RAPD profile based genetic characterization of chemotypic variants of Artemisia annua L. IUBMB Life 47:935–944
Sangwan NS, Kumar R, Srivastava S, Kumar A, Gupta A, Sangwan RS (2010) Recent developments on secondary metabolite biosynthesis in Artemisia annua L. J Plant Biol 37:1–24
Schramek N, Wang H, Römisch-Margl W, Keil B, Radykewicz T, Winzenhörlein B, Beerhues L, Bacher A, Rohdich F, Gershenzon J, Liu B, Eisenreich W (2010) Artemisinin biosynthesis in growing plants of Artemisia annua A 13 CO2 study. Phytochemistry 71:179–187
Singh N, Luthra R, Sangwan RS (1990) Oxidative pathways and essential oil biosynthesis in the developing Cymbopogon flexuosus leaf. Plant Physiol Biochem 28:703–710
Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS (2006) Artemisia annua L (Asteraceae) trichome-specific cDNAs reveal CYP71AV1, a cytochrome P450 with a key role in the biosynthesis of the antimalarial sesquiterpene lactone artemisinin. FEBS Lett 580:1411–1416
Towler MJ, Weathers PJ (2007) Evidence of artemisinin production from IPP stemming from both the mevalonate and the nonmevalonate pathways. Plant Cell Rep 26:2129–2136
VanNieuwerburgh FCW, VandeCasteele SRF, Maes L, Goossens A, Inze D, VanBocxlaer J, Deforce DLD (2006) Quantitation of artemisinin and its biosynthetic precursors in Artemisia annua L. by high performance liquid chromatography-electrospray quadrupole time-of-flight tandem mass spectrometry. J Chroma A 1118:180–187
Yadav RK, Sangwan RS, Sabir F, Sangwan NS (2013) Effect of prolonged water stress on specialized secondary metabolites, peltate glandular trichomes, and pathway gene expression in Artemisia annua L. Plant Physiol Biochem 74:70–83
Zeng MY, Li LN, Chen SF, Li GY, Liang XT, Chen M, Clardy J (1983) Chemical transformations of qinghaosu, A peroxidic antimalarial. Tetrahedron 39:2941–2946
Zhang Y, Teoh KH, Reed DW, Maes L, Goossens A, Olson DJ, Ross AR, Covello PS (2008) The molecular cloning of artemisinic aldehyde D11(13) reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J Biol Chem 283:21501–21508
Zhao SS, Zeng MY (1985) Studies on the analysis of qinghaosu by high-pressure liquid chromatography and spectrometry (HPLC). Planta Med 51:233–237
Acknowledgments
Authors are thankful to CSIR-SIMPLE (BSC 109) and DST, Government of India, New Delhi for providing the financial grant to conduct the studies. RK is thankful to UGC, N Delhi for senior research fellowship. Authors thank Dr R S Bhakuni for providing standards and Dr A K Gupta for providing seeds. Authors are grateful to Director CSIR-CIMAP for constant support and encouragement.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yadav, R.K., Sangwan, R.S., Srivastava, A.K. et al. Comparative profiling and dynamics of artemisinin related metabolites using efficient protocol and expression of biosynthetic pathway genes during developmental span of two elite varieties of Artemisia annua L. J. Plant Biochem. Biotechnol. 24, 167–175 (2015). https://doi.org/10.1007/s13562-013-0249-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-013-0249-z