
Abstract
The potent antimalarial sesquiterpene lactone, artemisinin, is produced in low quantities by the plant Artemisia annua L. The source and regulation of the isopentenyl diphosphate (IPP) used in the biosynthesis of artemisinin has not been completely characterized. Terpenoid biosynthesis occurs in plants via two IPP-generating pathways: the mevalonate pathway in the cytosol, and the non-mevalonate pathway in plastids. Using inhibitors specific to each pathway, it is possible to resolve which supplies the IPP precursor to the end product. Here, we show the effects of inhibition on the two pathways leading to IPP for artemisinin production in plants. We grew young (7–14 days post cotyledon) plants in liquid culture, and added mevinolin to the medium to inhibit the mevalonate pathway, or fosmidomycin to inhibit the non-mevalonate pathway. Artemisinin levels were measured after 7–14 days incubation, and production was significantly reduced by each inhibitor compared to controls, thus, it appears that IPP from both pathways is used in artemisinin production. Also when grown in miconazole, an inhibitor of sterol biosynthesis, there was a significant increase in artemisinin compared to controls suggesting that carbon was shifted from sterols into sesquiterpenes. Collectively these results indicate that artemisinin is probably biosynthesized from IPP pools from both the plastid and the cytosol, and that carbon from competing pathways can be channeled toward sesquiterpenes. This information will help advance our understanding of the regulation of in planta production of artemisinin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Overexploitation of natural sources for high-valued chemicals has increased interest in finding alternative production platforms to facilitate preservation of endangered species and the environment (Wu et al. 2006). Low concentrations of products in plants and expensive extraction processes motivate a deeper understanding of their complex synthesis. Terpenoids constitute the largest family of natural plant products with over 30,000 members (Sacchettini and Poulter 1997; Dewick 2002).
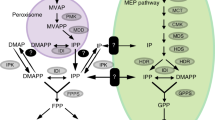
Until the discovery of an alternative non-mevalonate pathway (MEP) (Fig. 1), it was widely accepted that the formation of isopentenyl diphosphate (IPP), the 5-carbon isoprene precursor to terpenoids, occurred via the mevalonate pathway (MVA). Some of the earliest data in plants came from the study of [13C]-labeled glucose incorporation into ginkgolides (diterpenes) in Ginkgo biloba (Schwarz 1994), which showed that the resulting labeling pattern was incompatible with the mevalonate route of synthesis. Prior to such labeling studies, Bach and Lichtenthaler (1983) used mevinolin, a known 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR) inhibitor, to study plant growth, sterol formation, and pigment accumulation. They suggested that there was a separate mevalonate-producing pathway and HMGR in plastids. The other studies demonstrating mevinolin’s lack of inhibition on plastidic terpenoids raised questions on the involvement of the mevalonate pathway in their biosynthesis. While there is some debate over the existence of a plastidic HMGR (Dubey et al. 2003), the “alternative” production of IPP via the non-mevalonate pathway in plants was definitively elucidated by Rohmer et al. (1996). It is now known that in higher plants, two independent pathways located in separate intracellular compartments are involved in the biosynthesis of IPP: the cytosolic mevalonate pathway (MVA) and the plastidic non-mevalonate pathway (MEP).

Generalized terpenoid pathway showing routes to IPP, artemisinin, and points of inhibition for the three inhibitors used in this study. Inhibitors are shown in black boxes and the enzymes affected are shown in bold. Dotted lines indicate exchange of precursors between cellular compartments. HMG-CoA hydroxymethylglutaryl CoA, MEV mevinolin, HMGR HMG-CoA reductase, MVA mevalonic acid, DMAPP dimethylallyl diphosphate, IPP isopentenyl diphosphate, FPP farnesyl diphosphate, GGPP geranylgeranyl diphosphate, MIC miconazole, 14DM 14α-demethylase, G3P glyceraldehyde-3-phosphate, DXP deoxylulose 5-phosphate, DXR DXP reductoisomerase, FOS fosmidomycin; DXS DXP synthase, MEP methylerithritol 4-phosphate, GPP geranyl diphosphate, ABA abscisic acid
Studies by Adam and Zapp (1998) and Steliopoulis et al. (2002) showed that sesquiterpene-directed IPP carbon comes from both the MEP and the MVA pathways in members of the dicot angiosperms, e.g., chamomile and goldenrod, which are members of the Compositae and closely related to Artemisia annua. In contrast, in the monocot angiosperms (Maier et al. 1998) and the more primitive bryophytes (Adam et al. 1998), sesquiterpenes derive all their IPP carbon from the MEP pathway. Thus, in the dicots, labeling studies suggested the mixing of IPP produced by the two pathways that are located in different subcellular compartments.
To date it is not known if one (or both) of the pathways provides IPP for the biosynthesis of the important antimalarial therapeutic, artemisinin, in A. annua. Here we present evidence showing that like other members of the Compositae, A. annua also appears to use IPP from both the MEP and the MVA pathways in artemisinin production.
Materials and methods
Cultures and their maintenance
Seeds of A. annua (YU strain; Weathers et al. 1994) were sterilized as described by Wang and Weathers (2007). Ten young seedlings (two-cotyledon stage) were transferred to 50 mL Erlenmeyer flasks containing 5 mL autoclaved B5 medium (Gamborg et al. 1968) pH 5.7 with 3% w/v sucrose. Flasks were incubated on a rotary shaker at 100 rpm, 23 ± 2°C, under continuous cool white fluorescent light (∼40 μE m−2 s−1) for 7 or 14 days post cotyledon emergence (dpc) prior to transfer to experimental medium containing inhibitors. All experimental medium was filter-sterilized (0.22 μm) to eliminate variation in initial sugar profiles due to thermal degradation of sucrose and the resulting monosaccharides during autoclaving (Weathers et al. 2004).
Inhibitors
Mevinolin (MEV) is a highly specific inhibitor of HMGR responsible for the conversion of HMG-CoA to mevalonic acid (MVA), the first intermediate specific to the mevalonate pathway (Bach and Lichtenthaler 1983). Fosmidomycin (FOS) inhibits production of 2-C-methyl-d-erythritol-4-phosphate (MEP) from 1-deoxy-d-xylulose-5-P (DXP), catalyzed by the enzyme DXR (DXP reductoisomerase) (Rodrígues-Concepción et al. 2004). MEP is the first intermediate specific to the non-mevalonate pathway. While MEV also strongly inhibits sterol biosynthesis in higher plants, the biosynthesis of chlorophyll, carotenoids, and plastoquinones remains unaffected (Fig. 1). Miconazole (MIC) interferes with sterol biosynthesis by inhibiting sterol 14α-demethylases (Zarn et al. 2003).
Stock solutions of inhibitors and relevant solvents were filter sterilized (0.22 μm) into sterile containers and stored at 4°C. FOS (10 mM; a kind gift from Jomaa Pharmaka, GmbH, Giessen Germany) was dissolved in diH2O. To ensure solubility MEV (10 mM; Sigma M-2147 or its equivalent lovastatin, Axxora ALX-430-103-M050) was hydrolyzed to mevinolin salt using the method of Rodrígues-Concepción and Gruissem (1999). MIC (40 mM; Sigma M-3512) was dissolved in DMSO (Sigma D-8779). Aliquots of stock solutions were added to experimental medium (filter sterilized B5 medium with 3% (w/v) sucrose) to yield the desired final concentrations and placed on a rotary shaker for 1 day prior to transfer of plants. Plants were incubated in medium containing inhibitors or corresponding control solvents for 7 or 14 days. The concentrations used and incubation times were based on data from preliminary experiments that enabled us to obtain statistically significant results.
Biomass measurements
Plants were harvested and gently blotted on paper towels before separation into shoots and roots. Any tissue below the cotyledons was considered to be root tissue. After blotted fresh weights (FW) were recorded, tissue was placed into pre-weighed aluminum pans in a 60°C oven for approximately 1 week to determine dry weights (DW).
Artemisinin analysis
At least 20 mg of dry shoot tissue was homogenized in glass test tubes and extracted three times with 2 mL toluene for 30 min in an ultrasonic water bath. Although roots enhance artemisinin levels in shoots, roots of the native plant do not contain artemisinin (Ferreira and Janick 1996), so they were not extracted. Pooled extracts for each replicate were dried in test tubes in a 30°C water bath under a nitrogen stream. The Q260 derivative of artemisinin was analyzed via HPLC according to the method of Smith et al. (1997) using a C18 column and a mobile phase of 40% (v/v) methanol and 60% 0.01 M sodium phosphate buffer, pH 7.0. Artemisinin levels varied considerably between experiments and seed batch used, so all artemisinin results were subsequently normalized and then compared against untreated controls.
Data analyses
Data were analyzed using ANOVA followed by Tukey HSD post hoc tests. There was a minimum of three replicates for each condition, and experiments were repeated at least twice unless noted otherwise.
Results
In order to assess the role of the two IPP-producing arms of the terpenoid pathway in the production of artemisinin, we measured artemisinin production in the presence of three inhibitors: two, FOS and MEV, that affect regulatory steps leading to IPP, and one, MIC, that affects carbon allocation between sesquiterpenes and sterols post FPP. Both the changes in morphology of A. annua seedlings used in the experiments as well as their production of artemisinin were measured.
Plant morphology
After several days only young (7 dpc) A. annua plants that were exposed to 100 μM FOS became chlorotic (see comparative composite photo in Fig. 2); this effect, however, was much less pronounced when older (14 dpc) plants were used as inocula. The pattern of chlorosis was interesting in that only leaves that emerged after exposure to FOS were affected with the yellowing progressing outward while leaf margins remained green (Fig. 3). While plants exposed to FOS looked healthy besides the chlorosis, the shoot and root biomass (DW per plant) did not significantly change compared to controls until plants were exposed to inhibitor for 14 days (Fig. 2). After 14 days overall growth declined by about 25%, and this occurred equally in both shoots and roots (Fig. 2). In contrast, both the 7 dpc and 14 dpc plants incubated with 100 μM MEV for any length of time had visibly stunted shoots that were slightly chlorotic, and had short, thick roots (Fig. 2). However, compared to the controls, the shoot and root mass per plant were not statistically different (Fig. 2).

Photos of 21-day-old A. annua seedlings exposed for 7–14 days treatment with 0.1% (v/v) H2O (control (a)), 100 μM FOS (b), and 100 μM MEV (c) in B5 medium with 3% (w/v) sucrose. Table shows correlating biomass changes, and letters within each row indicate statistical significance at P = 0.05. Lack of statistically significant change in FOS + MEV roots at 14 days is due to only three samples in the data pool

Photos of 21-day-old A. annua seedlings exposed for 7–14 days to 100 μM FOS. Note the pattern of chlorosis
Plants exposed to DMSO (0.5% v/v) containing 200 μM MIC for 14 days exhibited only slight chlorosis. Further, the MIC-grown plants were only slightly more yellow than the DMSO controls (Fig. 4). In contrast to the pattern of chlorosis observed in FOS-grown seedlings, the margins of older leaves in MIC-grown seedlings turned yellow. Most plants, including the untreated and DMSO controls showed some visible degree of yellowing after 14 days. There was no statistically significant effect of MIC on shoot or root DW versus either set of controls (H2O or DMSO).

Photos of 21-day-old A. annua seedlings exposed for 7–14 days treatment with 0.1% (v/v) H2O (control (a)), 200 μM MIC (b), and 0.05% (v/v) DMSO (c) in B5 medium with 3% (w/v) sucrose. Table shows correlating biomass changes, and letters within each row indicate statistical significance at P = 0.05
Artemisinin production
Despite many attempts, we were unable to obtain a single clone of A. annua (YU strain) shoot cultures to use in these studies; thus, each replicate sample contained groups of 10 seedlings that were pooled for artemisinin analysis in order to normalize variation in artemisinin production for the genetic pool. Additionally, data are expressed as amount produced per total shoot DW relative to control plants since artemisinin content in a batch of seedlings can vary considerably (0.6–8.4 μg g DW−1) from experiment to experiment. Although not understood, this variation in artemisinin content seemed to correlate with the batch of seeds used in each experiment.
Using this experimental approach, FOS and MEV each significantly decreased the production of artemisinin, whereas application of both inhibitors completely inhibited production of artemisinin (Fig. 5). Indeed we observed that complete inhibition of artemisinin production occurred after only 7 days incubation in both inhibitors. While exposure to either FOS or MEV decreased artemisinin production compared to control plants, it was only consistent and statistically significant for young (7 days) plants exposed to inhibitors for 14 days (Fig. 5). We therefore hypothesized that the reduced effects on morphology and artemisinin production in older plants might be due to the greater amount of biomass present compared to the amount of inhibitor provided. However, proportionately increasing the concentration of inhibitors to parallel the increase in biomass of older plants did not result in statistically significant changes in artemisinin production. We can thus infer that it is likely the developmental stage of the plant that plays a role in its response to the inhibitors and younger plants are more susceptible to the stress of inhibition both physiologically and biochemically.

Change in artemisinin (AN) content in shoots of 21-day-old A. annua seedlings exposed for 7–14 days treatment with 0.1% (v/v) H2O (control), 100 μM FOS, 100 μM MEV, and 100 μM FOS + MEV in B5 medium with 3% (w/v) sucrose. Data from FOS + MEV not shown because artemisinin content was zero. Letters indicate statistical differences at P = 0.05
Carbon channeling
Experiments with the sterol inhibitor MIC showed that when young (7 days) seedlings were exposed to MIC for 14 days they produced up to 11.5 times more artemisinin than controls (Fig. 6). Since MIC is not water soluble, it was dissolved in DMSO, and we ran additional control cultures containing DMSO without MIC. Interestingly, it appears that DMSO itself stimulates artemisinin production (up to six times more than controls). As observed for FOS and MEV-grown seedlings, the age of the plants appears to influence the response. Results from experiments using older seedlings were not as definitive or reproducible, but this approach for increasing artemisinin production certainly bears further investigation, especially since effects on plant health appeared to be minimal.

Change in artemisinin (AN) content in shoots of 21-day-old A. annua seedlings exposed for 7–14 days treatment with 0.05% (v/v) H2O (control), 200 μM MIC, and 0.05% (v/v) DMSO in B5 medium with 3% (w/v) sucrose. Letters indicate statistical differences at P = 0.05
Discussion
Clearly the gold standard for asking a question on carbon distribution between these pathways would be experimenting using [1-C] labeled glucose, followed by analysis of the carbon labeling patterns in isolated artemisinin or its post FPP precursors. Unfortunately, we were unable to obtain adequate enrichment of artemisinin or its immediate precursors despite using many different approaches for incorporation of a heavy label. Subsequently we resorted to using inhibitors specific to each pathway.
Plants exposed to FOS exhibited chlorosis, which was anticipated due to inhibition of the pathway leading to chlorophylls (Fig. 1). Chlorosis was exceptionally rapid and severe in young plants, which may explain the slight decrease in plant biomass versus controls. Since only newly developing leaves exhibited chlorosis, and the yellowing occurred from the basal to apical direction (Fig. 3), this suggested that the older tissue was less susceptible to the negative effects of FOS. Indeed Heintze et al. (1990) showed that mature chloroplasts had a much higher incorporation rate of labeled mevalonate into plastidic isoprenoids than developing chloroplasts, suggesting that mature chloroplasts have mechanisms whereby they are better able to compensate for deficiencies in precursor levels, specifically the ability to import IPP from the mevalonate pathway. Similarly Hemmerlin and Bach (1998) noted that young tobacco cells were more sensitive to MEV than older ones in terms of cell death. Exposure of A. annua plants to MEV tended to promote stunted shoots that were slightly chlorotic and short, thickened roots. These effects have also been observed by others (Re et al. 1995; Kasahara et al. 2002; Rodrígues-Concepción et al. 2004).
While we can reasonably conclude from our data that both pathways are probably involved in artemisinin production, various tracer studies have shown that the compartmental separation of the two pathways is not always absolute (Arigoni et al. 1999; Theil and Adam 2002), so the extent to which each pathway contributes to the production of artemisinin is not known. Both operate simultaneously, and several studies have indicated that the two pathways can cooperate and exchange various precursors (Hemmerlin et al. 2003; Rodrígues-Concepción et al. 2004 and references therein; Dudareva et al. 2005). Exposing A. annua seedlings to FOS and MEV simultaneously severely reduced their growth (Fig. 2). Bartram et al. (2006) showed that in lima beans grown under similar conditions all de novo terpenoid biosynthesis was suppressed.
The possible transport of IPP from one compartment to the other could pose another interpretation of our data. One might consider that the decrease in artemisinin in the presence of inhibitors may be a consequence of the presumably very low levels of IPP in one compartment or the other after 2 weeks of inhibition. For example, in the presence of FOS, IPP in the plastids decreases dramatically and IPP could thus be transported from cytoplasm to plastid to compensate for the loss. Imported cytoplasmic IPP might then be used to make critical plastid components resulting in a decrease in artemisinin. However, our results do not support this premise. If in FOS, IPP moved from cytosol to plastid, then one would expect the plastids to be able to maintain their pigmentation. They do not. Chlorophyll bleaches quite noticeably within 48 h, and the plantlets become pale (Fig. 3). Both chlorophylls and carotenoids are made in the plastids from IPP/DMAPP → GGPP (Fig. 1). The bleaching response suggests that any IPP made in the cytosol is either not adequate to compensate for the loss of IPP in the plastid, or that IPP is not being moved from cytosol to plastid. Laule et al. (2003) showed a similar response to FOS in Arabidopsis, and when they measured the labeled metabolites of both cytosol and plastids, IPP compensation ability was much more pronounced going from plastid to cytosol than from cytosol to plastid.
Although MIC and its solvent, DMSO, did not affect plant growth, MIC significantly increased artemisinin yields compared to either the water or the DMSO controls (Figs. 4, 6) confirming the earlier work of Kudakasseril et al. (1987). Many enzymes involved in plant sterol biosynthesis are encoded by multiple genes (Bach and Benveniste 1997) and the CYP51 gene that encodes the 14α-demethylases inhibited by MIC may indeed have several genes in A. annua. Arabidopsis has two, and rice has 10 (Kim et al. 2005), some of which show differential growth and tissue expression patterns. While we did not analyze the sterol content of MIC-treated plants, the lack of visible tissue defects due to sterol inhibition may have been due to multiple genes functioning together.
The stimulatory effect of DMSO on artemisinin production was unexpected. Others have reported that DMSO can promote phage transcription (Chen and Zhang 2005), and can alter other cellular functions (Santos et al. 2003). In plants DMSO was reported to modulate Zn uptake, respiration, and RNA and protein metabolism in bean tissues (Bajaj et al. 1970), and stimulate Hibiscus protoplast division (Carswell et al. 1989). Other plant species were, however, either inhibited (Hahne and Hoffman 1984), or not affected by the chemical (Carswell et al. 1989). Thus, the effects of DMSO on secondary metabolism in plants should be further studied in order to determine its mechanism of action and possible utility in overproduction of valuable secondary metabolites like artemisinin.
In summary, we have shown that both the cytosol-based mevalonate pathway and the plastid-based non-mevalonate pathway are likely involved in the production of IPP for artemisinin biosynthesis. The proportion of IPP supplied by each pathway, however, is unknown since some metabolites may be exchanged between the two compartments, making it impossible to track their origin without the assistance of additional tools such as tracers. Younger plants were more susceptible to the inhibitors than older plants, and increasing the concentration of inhibitors proportional to the increase in biomass did not result in a corresponding inhibitory response. Although not yet understood, it appears that more mature plants are better able to compensate against metabolic inhibition. We were also able to increase artemisinin production by inhibiting sterol biosynthesis, and these results were again more striking in younger plants. We also showed that DMSO had a significant stimulatory effect on artemisinin production. Taken together these data will be useful in helping us to understand better the pathways leading to artemisinin production with a longer term intent of increasing yields of this important therapeutic in planta.
References
Adam K, Zapp J (1998) Biosynthesis of the isoprene units of chamomile sesquiterpenes. Phytochem Anal 48:953–959
Adam K, Thiel R, Zapp J, Becker H (1998) Involvement of the mevalonic acid pathway and the glyceraldehyde-pyruvate pathway in terpenoid biosynthesis of the liverworts Ricciocarpos natans and Conocephalum conicum. Arch Biochem Biophys 354:181–187
Arigoni D, Eisenreich W, Latzel C, Sagner S, Radykewicz T, Zenk MH, Bacher A (1999) Dimethylallyl pyrophosphate is not the committee precursor of isopentenyl pyrophosphate during terpenoid biosynthesis from 1-deoxyxylulose in higher plants. Proc Natl Acad Sci USA 96:1309–1314
Bach TJ, Benveniste P (1997) Cloning of cDNAs or genes encoding enzymes of sterol biosynthesis from plants and other eukaryotes: heterologous expression and complementation analysis of mutations for functional characterization. Prog Lipid Res 36:197-226
Bach TJ, Lichtenthaler HK (1983) Inhibition by mevinolin of plant growth, sterol formation and pigment accumulation. Physiol Plant 59:50–60
Bajaj YPS, Rathore VS, Wittwer SH, Adams MW (1970) Effect of dimethyl sulfoxide on zinc65 uptake, respiration, and RNA and protein metabolism in bean (Phaseolus vulgaris) tissues. Am J Bot 57:794–799
Bartram S, Jux A, Gleixner G, Boland W (2006) Dynamic pathway allocation in early terpenoid biosynthesis of stress-induced lima bean leaves. Phytochem 67:1661–1672
Carswell GK, Johnson CM, Shillito RD, Harms CT (1989) O-acetyl-salicylic acid promotes colony formation from protoplasts of an elite maize inbred. Plant Cell Rep 8:282–284
Chen Z, Zhang Y (2005) Dimethyl sulfoxide targets phage RNA polymerases to promote transcription. Biochem Biophys Res Commun 333:664–670
Dewick PM (2002) The biosynthesis of C5-C25 terpenoid compounds. Nat Prod Rep 19:181–222
Dubey VS, Bhalla R, Luthra R (2003) An overview of the non-mevalonate pathway for terpenoid biosynthesis in plants. J Biosci 28:637–646
Dudareva N, Andersson S, Orlova I, Gatto N, Reichelt M, Rhodes D, Boland W, Gershenzon J (2005) The nonmevalonate pathway supports both monoterpene and sesquiterpene formation in snapdragon flowers. Proc Natl Acad Sci USA 102:933–938
Ferreira JFS, Janick J (1996) Roots as an enhancing factor for the production of artemisinin in shoot cultures of Artemisia annua. Plant Cell Tissue Organ Cult 44:211–217
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:148–151
Hahne G, Hoffmann F (1984) Dimethyl sulfoxide can initiate cell divisions of arrested callus protoplasts by promoting cortical microtubule assembly. Proc Natl Acad Sci USA 81:5449–5453
Heintze A, Gorlach J, Leuschner C, Hoppe P, Hagelstein P, Schulze-Siebert D, Schultz G (1990) Plastidic isoprenoid synthesis during chloroplast development. Plant Physiol 93:1121-1127
Hemmerlin A, Bach TJ (1998) Effects of mevinolin on cell cycle progression and viability of tobacco BY-2 cells. Plant J 14:65–74
Hemmerlin A, Hoeffler JF, Meyer O, Tritsch D, Kagan IA, Grosdemange-Billiard C, Rohmer M, Bach T (2003) Cross-talk between the cytosolic mevalonate and the plastidial methylerythritol phosphate pathways in tobacco bright yellow-2 cells. J Biol Chem 278:26666–26676
Kasahara H, Hanada A, Kuzuyama T, Takagi M, Kamiya Y, Yamaguchi S (2002) Contribution of the mevalonate and methylerythritol phosphate pathways to the biosynthesis of gibberellins in Arabidopsis. J Biol Chem 277:45188–45194
Kim HB, Schaller H, Goh CH, Kwon M, Choe S, An CS, Durst F, Feldmann KA, Feyereisen R (2005) Arabidopsis cyp51 mutant shows postembryonic seedling lethality associated with lack of membrane integrity. Plant Physiol 138:2033–2047
Kudakasseril GJ, Lam L, Staba EJ 1987 Effect of sterol inhibitors on the incorporation of 14C-isopentenyl pyrophosphate into artemisinin by a cell-free system from Artemisia annua tissue cultures and plants. Planta Med 53:280–284
Laule O, Fürholz A, Chang HS, Zhu T, Wang X, Heifetz, Gruissem W, Lange BM (2003) Crosstalk between cytosolic and plastidial pathways of isoprenoid biosynthesis in Arabidopsis thaliana. Proc Natl Acad Sci USA 100:6866–6871
Maier W, Schneider B, Strack D (1998) Biosynthesis of sesquiterpenoid cyclohexenone derivatives in mycorrhizal barley roots proceeds via the glyceraldehyde 3-phosphate/pyruvate pathway. Tetrahedron Lett 39:521–524
Re EB, Jones D, Learned RM (1995) Co-expression of native and introduced genes reveals cryptic regulation of HMG CoA reductase expression in Arabidopsis. Plant J 7:771–784
Rodrígues-Concepción M, Gruissem W (1999) Arachidonic acid alters tomato HMG expression and fruit growth and induces 3-hydroxy-3-methylglutaryl coenzyme A reductase-independent lycopene accumulation. Plant Physiol 119:41–48
Rodrígues-Concepción M, Fores O, Martinez-Garcia JF, Gonzalez V, Phillips MA, Ferrer A, Boronat A (2004) Distinct light-mediated pathways regulate the biosynthesis and exchange of isoprenoid precursors during Arabidopsis seedling development. Plant Cell 16:144–156
Rohmer M, Seemann M, Horbach S, Bringermeyer S, Sahm H (1996) Glyceraldehyde 3-phosphate and pyruvate as precursors of isoprenic units in an alternative non-mevalonate pathway for terpenoid biosynthesis. J Am Chem Soc 118:2654–2566
Sacchettini JC, Poulter CD (1997) Creating isoprenoid diversity. Science 277:2788–1789
Santos NC, Figueira-Coelho J, Martins-Silva J, Saldanha C (2003) Multidisciplinary utilization of dimethyl sulfoxide: pharmacological, cellular, and molecule aspects. Biochem Pharmacol 65:1035–1041
Schwarz KM (1994) Terpen-biosynthese in Ginkgo biloba: Eine Uberraschende geschichte, Ph.D. Thesis, Eidgenossischen Technischen Hochschule, Zurich, Switzerland
Smith TC, Weathers PJ, Cheetham RD (1997) Effects of gibberellic acid on hairy root cultures of Artemisia annua: growth and artemisinin production. In Vitro Cell Dev Biol Plant 33:75–79
Steliopoulos P, Wüst M, Adam K-P, Mosandl A (2002) Biosynthesis of the sesquiterpene germacrene D in Solidago cnadensis: 13C and 2H labeling studies. Phytochem Anal 60:13–20
Thiel R, Adam KP (2002) Incorporation of [1–13C]1-deoxy-d-xylulose into isoprenoids of the liverwort Conocephalum conicum. Phytochem Anal 59:269–274
Wang Y, Weathers PJ (2007) Sugars proportionately affect artemisinin production. Plant Cell Rep 26:1073–1081
Weathers PJ, Cheetham RD, Follansbee E, Teoh K (1994) Artemisinin production by transformed roots of Artemisia annua. Biotechnol Lett 16:1281–1286
Weathers PJ, DeJesus-Gonzalez L, Kim YJ, Souret FF, Towler MJ (2004) Alteration of biomass and artemisinin production in A. annua hairy roots by media sterilization method and sugars. Plant Cell Rep 23:414–418
Wu S, Schalk M, Clark A, Miles RB, Coates R, Chappell J (2006) Redirection of cytosolic or plastidic isoprenoid precursors elevates terpene production in plants. Nat Biotechnol 24:1441–1447
Zarn JA, Brüschweiler BJ, Schlatter JR (2003) Azole fungicides affect mammalian steroidogenesis by inhibiting sterol 14α-demethylase and aromatase. Environ Health Perspect 111:255–261
Acknowledgments
Thanks to Brandon Matthews for some preliminary work. We also appreciate assistance with statistics by Professor Liz Ryder of WPI, and the critical reviews provided by Professors Kris Wobbe at WPI, and Fabricio Medina-Bolivar at the Arkansas Bioscience Institute. Financial support for this work was gratefully received from NIH 1R15GM 069562-01.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Lakshmanan.
Rights and permissions
About this article
Cite this article
Towler, M.J., Weathers, P.J. Evidence of artemisinin production from IPP stemming from both the mevalonate and the nonmevalonate pathways. Plant Cell Rep 26, 2129–2136 (2007). https://doi.org/10.1007/s00299-007-0420-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-007-0420-x