
Abstract
A set of 24 genotypes bred at different centres in India as well as in CIMMYT showing variability for drought tolerance were selected for molecular and morpho-physiological characterization. A set of 35 SSR markers, having genome-wide coverage, was chosen for genotyping the inbreds. These markers generated a total of 111 polymorphic alleles with an average of 3.17 alleles per locus. The minimum and maximum PIC value was 0.27 and 0.77 with a mean of 0.5. A total of 13 unique alleles were found in the 24 inbred lines. The coefficient of genetic dissimilarity ranged from 0.192 to 0.803. NJ-based tree suggested the presence of three major clusters of which, two of them had subgroups. Phenotyping of inbreds by morpho-physiological traits revealed that there was a positive relationship among root length, chlorophyll content, relative water content while anthesis-silking interval was negative relationship with all these traits. Genotyping data complemented by morpho-physiological parameters were used to identify a number of pair-wise combinations for the development of mapping population segregating for drought tolerance and potential heterotic pairs for the development of drought tolerant hybrids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The importance of maize (Zea mays L.) in Indian farming system is very much realized and the crop is gaining more popularity in recent days due its multiple uses. The crop is next to pearl millet and sorghum with respect to drought tolerance. Traditionally, in India, maize is grown in kharif but it now finds a place in rabi by replacing other rabi crops. Nearly 85% of the total area is now under kharif cultivation, while rabi maize occupies <10% of the area and contributes more than 25% of the annual production (Singh et al. 2005). Both kharif and rabi seasons are prone to drought stress at the critical stages of the crop growth particularly at pre-flowering stage. Drought in kharif is because of delay or failure of monsoon rain whereas in rabi, it is due to non availability of irrigation water during March-April which normally coincides with flowering and grain filling stages. Grain yield under drought stress affects the grain yield dramatically. Development of drought tolerant hybrids is the only approach to mitigate the challenges posed by climate change and the ever decreasing irrigation water to the Indian cropping systems.
Knowledge on genetic diversity in crop plants is very important to understand the trait variability. Genetic relationship of maize germplasms was characterized with the help of morphological traits as early as in 70’s (Goodman and Bird 1977). Nowadays, morphological characters are less preferred for genetic diversity studies because they are limited in number, environmentally influenced, less polymorphic, stage specific expression, low heritability and various other reasons (Smith and Smith 1992). On the other hand, DNA markers are used in genetic characterization and diversity studies since they are devoid of the above-mentioned limitations (Saker et al. 2005; Souza et al. 2008). Among many marker systems, simple sequence repeats (SSRs) are considered suitable because of their abundance in the genome (Matsuoka et al. 2002), high variability (Tautz 1989; Schug et al. 1998), uniform distribution in the genome (Liu et al. 1996; Senior et al. 1996). SSRs are PCR-based, codominant in inheritance and multiallelic marker system (Chin et al. 1996). The assay is less laborious and inexpensive since the alleles can be separated by even simple agarose gel electrophoresis (Senior et al. 1998; Pinto et al. 2003).
Several studies have extensively used molecular markers to assess the genetic relationship of genotypes in maize (Dubreuil et al. 1996; Smith et al. 1997; Ajmone-Marsan et al. 1998; Senior et al.1998; Dubreuil and Charcosset, 1999; Lu and Bernardo 2001; Enoki et al. 2002; Gethi et al. 2002; Legesse et al. 2007; Van Inghelandt et al. 2010), in ground nut (Jiang et al. 2010), in sorghum (Dickson et al. 2011) and in wheat (Chenyang et al. 2011). Many studies used SSR markers in maize (Lu and bernardo 2001; Vigouroux et al. 2002; Warburton et al. 2002; Liu et al. 2003; le Clerc et al. 2005; Warburton et al. 2008) since they detect high levels of polymorhphism (Smith et al. 1997), are amenable for automation (Sharon et al. 1997) and, provide precision and reproducible assays (Heckenberger et al. 2002).
Identification of heterotic pairs to improve agronomic traits from genetic diversity studies has been seen in many crops. Melchinger and Gumber (1998) discussed the possibilities of establishment of heterotic pools in maize using genetic distance based on molecular markers. The ability of SSR markers to identify the potential heterotic groups in maize has been demonstrated in previous studies (Enoki et al. 2002; Reif et al. 2003; Barata and Carena, 2006). The inclusion of exotic germplasm between the groups has also been suggested to increase the genetic difference thereby improving the heterotic potential among the groups (Beck et al. 1991; Ron Parra and Hallauer 1997).
Development of a mapping population to map QTLs for drought is a big investment in breeding program hence, selection of an appropriate parental pair should be given much importance. Molecular markers to a certain extent can help in assessment of genetic distance and the level of polymorphism among the given genotypes but genotypic data combined with morpho-physiological data would be a good option to choose the parents. Morpho-physiological traits, which can be quantified and realized visually, play major role in selection of parental lines especially for developing population to map drought QTLs.
The present experiment was carried out with the following objectives: to characterize the maize inbred lines using SSR markers and morpho-physiological traits, to assess the genetic relationship of the inbred lines, and to find suitable parental pairs for developing mapping population segregating for putative drought QTLs.
Materials and methods
Materials and genotyping
A total of 24 inbred lines, representing Indian and CIMMYT collections were used in the present study for molecular characterization (Table 1). These genotypes were grown in individual cups and the leaf sample from 10 to 15 seedlings was harvested in bulk after removing the midrib and leaf tip from 14-day old plants. Total genomic DNA was isolated by using CTAB procedure (Murray and Thompson, 1980). Based on quantification results a working stock of 20 ng/ul was prepared using the mother stock.
A total of 50 SSR markers with good coverage on the maize genome, were initially selected for genotyping and the PCR was carried out in 10 μl reaction mixture containing 2 ul of 20 ng/ul genomic DNA as template, 2 mM MgCl2, 1 mM dNTPs, 2 μM primer pair (forward and reverse) and 1.5 U Taq Polymerase (GeNei, Mumbai). The amplification was carried out with initial denaturation of 94°C for 3 min and followed by a “touch-down” procedure with two steps. The first step had 12 cycles: denaturation at 94°C for 30 s, annealing at 63°C for 30 s (subsequently the annealing temperature was reduced by 0.5°C per cycle), and extension at 72°C for 45 s. The second step was set for 35 cycles: denaturation at 94°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 45 s. The extension step was carried out at 72°C for 7 min. The resulting PCR amplicons were resolved in 3.5% agarose gel at 120 V for 3–4 h. The amplicons were scored as alleles for the loci. The alleles were scored manually and allele sizes (base pairs) were determined comparing with 100 bp DNA ladder which is run parallel with the inbred lines.
Phenotyping
The 24 maize inbred lines were evaluated for their response to drought during kharif 2010 at the Maize Research Farm of the Indian Agricultural Research Institute, New Delhi (Table 1). Soil of the experimental plots was sandy loam with pH 7.8. The set of 24 lines were evaluated under well-water and water stressed situations in RBD with two replications. Each entry was planted in single row of 1.75 m long, with 0.20 m spacing within and 0.60 m between rows. All the entries were sown during the 2nd of July 2010 and thinned to one plant per hill at the V2 growth stage. The crop was raised following all recommended package of practices except that nitrogen was applied in 5 split doses (basal, seedling, knee high, flowering and grain filling stages).
The target stage for imposing drought was the reproductive phase, including flowering (both male and female), pollination, fertilization and early grain filling stage (lag phase, which is known to be completely dependent on current assimilate supply). The drought stress was imposed under managed stress (rain-out shelter) conditions by withdrawing irrigation and closing the top of rain-out shelter 2 weeks before 50% anthesis till 2 weeks after completion of 50% silking. After the completion of drought treatment the top of the rain-out shelter was moved to its platform.
Morpho-physiological observations were made for anthesis-silking interval (ASI), chlorophyll content (CC), leaf area index (LAI), root length (RL), and relative water content (RWC). For measuring root length, the plants were dug out carefully. Roots were separated from the plant and washed in running tap water for complete removal of sand particles. The RL was measured by spreading root system and expressed in cm. The CC was measured with SPAD Chlorophyll meter (Minolta, Japan, Model 502) and expressed in SPAD value. The RWC (in%) of leaf was measured by the method described by (Smart and Bingham 1974). The LAI was measured by plant canopy analyzer (LICOR Bioscience, USA, Model LAI 2200). The ASI (in days) was calculated by subtracting days to 50% anthesis from days to 50% silking.
Data analysis
After the initial screen, 15 monomorphic and poorly amplified SSR markers were rejected from the experiment. The allele size data from 35 markers were collected for further statistical analysis. Major allele frequency, allele number, gene diversity, heterozygosity and polymorphic information content (PIC) were estimated using PowerMarker V3.0 (Liu and Muse 2005). Genetic dissimilarity was calculated by simple matching coefficient and the phylogenetic tree was constructed by Neighbour Joining (weighted average) method implemented in DARwin-5.0 (Perrier et al. 2003). Replicated data of morpho-physiological were subjected to a linear mixed model using ReML in GENSTAT (GENSTAT 14 Committee, 2011). Spearman’s rank correlation among the traits was done using the same software.
Results and discussion
The summary statistics of marker related parameters are presented in Table 2. Of the 35 SSR markers selected, the number of di-, tri- and tetra-nucleotide repeats was 22, 10 and 1, respectively. One SSR marker (bnlg125) belonged to compound repeat and one was a probe (umc1937). Markers with tri-nucleotide repeats are robust and more useful for assessing the genetic diversity since the variation in tri-nucleotide repeats are originated from the protein coding region (Tóth et al. 2000). The selected SSR markers uniformly covered all the 10 chromosomes and seven markers out of 35 were located on chromosome 5.
The 35 polymorphic SSR markers produced as many as 111 alleles with an average of 3.17 alleles per locus in the 24 genotype panel. The average number of alleles per loci (2 to 8) obtained in the present study was higher considering the number of genotypes examined in this study. The average number of alleles per locus was in accordance with the previous report (Pinto et al. 2003).
The allele size ranged from 80 bp (umc2174 and umc1761) to 340 bp (bnlg125). The number of alleles per locus varied from 2 (umc1552, umc1690¸ umc1325, umc1761, umc2201, umc2358 and umc2393) to 8 alleles (umc1056). umc1325 had the major allele frequency of 0.82 for allele 125 bp, indicated that it occurred 82% in the panel. Whereas allele 170 bp from umc1056 having the minimum value of 0.33 showed that it occurred only 33% in the panel. The low value of major allele frequency indicated that the selected genotype panel was highly diverse for the locus.
The level of heterozygosity in the inbred panel was significantly low. The mean value of heterozygosity was 0.16 implying that most of the loci attained homozygosity. However, for the loci umc2174, umc1056 and umc2341, the heterozygosity was 0.35. The presence of heterozygosity in the inbred lines might be due to a number of probable causes including residual heterozygosity, pollen or seed contamination, mutation at specific SSR loci, or amplification of similar sequences in different genomic regions due to duplication (Semagn et al. 2006). Since maize is a highly cross-pollinated crop, the most plausible explanation could be the residual heterozygosity which is not uncommon in maize. By virtue of its cross pollinated nature, maize maintains 5–10% heterozygosity which is called residual heterozygosity. As a result of this, inbreds tend to segregate for a few loci/characters despite repeated cycles of selfing over many generations. However, the low level of heterozygosity in the inbred lines revealed that they have been maintained properly and the reported heterozygosity was inherent.
The mean PIC value was nearly 0.5, which was sufficient to group the population into different clusters. Locus umc1552 had the lowest PIC value (0.27) whereas locus umc1056 had the highest PIC value (0.77). The PIC value will in fact, depend upon the germplasm that was under investigation. Closely related lines would express lower PIC, where as genetically diverse lines exhibit higher PIC values. Apart from being a different marker system which can detect polymorphisms, the ability to resolve the alleles also plays a crucial role in detecting the number of alleles. Though we used agarose to resolve the alleles, the mean PIC value was comparable with the previous findings (Senior et al. 1998; Heckenberger et al. 2002; Vaz Patto et al. 2004), because of the nature markers that we have chosen for polymorphism. The alleles of different markers gave clear cut banding pattern and separated well from each other.
Unique alleles are the alleles present only in a particular genotype and absent in other genotypes. A total of 13 unique alleles were identified in 35 markers. umc1056 generated a maximum of three unique alleles (120 bp in LM 14, 150 bp in DTPYC9-F46-3-6 and 185 bp in HKI 335) followed by umc2298 which generated two unique alleles (140 bp in CM139 and 150 bp in CML69). Markers umc2165, umc2226, umc2061, bnlg1702, umc2017 and phi087 had one unique allele with a size of 210 bp (CM 151), 135 bp (PC 3), 200 bp (DTPYC9-F46-3-6), 225 bp (HKI 335), 150 bp (DTPYC9-F46-3-6) and 180 bp (CML69), respectively. Further exploration of these unique alleles is very important since they would often associate with trait expression and will be useful in fingerprinting.
Assessment of genetic variability among the inbreds is a prerequisite for a multitude of benefits including protection of genetic identity such as intellectual property rights, conservation of genetic resources, and identification of potential heterotic groups and development of mapping populations. In the present study, we have assessed genetic diversity of the selected inbred lines for development of mapping populations segregating for putative drought QTLs in different agro-ecological zones. The genetic dissimilarity calculated based on simple matching coefficient was ranged from 0.192 to 0.803 with a mean of 0.536. The wider coefficient values in the inbred panel suggested that the genotypes were highly dissimilar. The mean genetic diversity was relatively high indicating that the SSR markers revealed high level of polymorphisms in the inbreds. The genetic diversity of our inbred panel revealed by SSR markers was very close to the findings of earlier results (Smith et al. 1997; Senior et al. 1998; Vaz Patto et al. 2004).
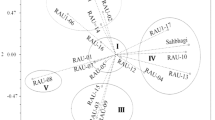
The weighted NJ method grouped the 24 genotypes into three major groups (a, b and c, Fig. 1). The group A had 11 genotypes and it was further divided into five subgroups (A-1, A-2, A-3, A-4 and A-5), of which A-2 and A-5 formed single genotype clusters. The second group (B) had two genotypes and the third group (C) had 11 genotypes. Group C was further separated into four subgroups (C-1, C-2, C-3 and C-4). Genotype LM 14 formed single genotype group (C-2) under group C. The markers separated the genotypes into different clusters and closely related genotypes clustered according to their pedigree. In group A, three genotypes (HKI 1532, HKI 577 and HKI 355) were grouped together (A-4) since they shared common pedigree. The DTPYC9 series had four genotypes and they formed a single sub-group (C-1) under group C. These four lines were bred at CIMMYT for drought tolerance. Similarly, pool 16 had three genotypes, derived from same pedigree found to be grouped in C-4. CM 139 and CM 140 formed independent sub-groups, indicating that they were distantly related from other genotypes of group A. The genetic dissimilarity of CM 139 and CM 140 had been already exploited in the heterosis breeding program resulted the release of hybrid “Parkash”. Our study reinstated the fact that these two were distantly related. LM 14 in group C also formed single cluster since it was genetically diverse from other members of group C. Group B had only two genotypes and they grouped together but separated from other two groups and they were susceptible to drought stress.

Clustering pattern of inbreds based on Weighted Neighbor Joining method. Major groups are denoted by a, b and c and the subgroups are indicated by numbers under the major group. Further subgroups are classified as T Tolerant, MT Moderately Tolerant, and S Susceptible based on their response to drought stress
This study opened up a lot of possibility of development of hybrids tolerance to drought stress by exploiting the heterosis. Since our experiment proved that the hybrid “Parkash” had the product of genetically dissimilar parental lines, the SSR-based clustering and the relationship among parental lines can be further used for development of new hybrids.
Significant variation was found in all the traits used in this experiment under stress condition (Table 3). A positive correlation was observed among root length, chlorophyll content, LAI and RWC where as ASI had negative relationship with other traits (Table 4). Longer roots will absorb more water so the RWC will go high. Broader leaf will hold more moisture so it will increase the chlorophyll content. These four traits had negative relationship with ASI which is again considered to be a good combination for breeding drought tolerance (Fig. 2). Since shorter ASI will ensure better pollination and escape from drought stress. All these traits complement to each other in mitigating drought stress. The morpho-physiological traits in conjunction with the genetic distance were used to short list the parental genotypes for development of mapping population and subsequent QTL mapping.

Mean performance of inbred lines under moisture stress condition. ASI, RL, CC and RWC are given in primary axis. LAI is given in secondary axis. Refer materials and methods for trait names
Lines from three distinct groups emerged as potential-drought tolerant lines based on phenotyping data. Two late maturity genotypes (HKI 577 and HKI 1752) from A-4 cluster, two genotypes (DTPWC9-F119-1-2 and DTPYC9-F46-3-6) from C-1 cluster and three genotypes (POOL 16 BNSEQ.C3 F32 × 37–4, POOL 16 BNSEQ.C3 F34 × 31–2, POOL 16 BNSEQ.C3 F10 × 36–2) from C-4 cluster were all drought tolerant genotypes and clear favorites for mapping population development under stress condition. The genotypes showed one- or two-day difference between anthesis and silking which is considered to be one of the important traits for breeding drought tolerance. Among them, genotypes HKI 577 and HKI 1532 were the top ranked inbreds having desirable characteristics. The value for root length, chlorophyll content and leaf area index of these two inbred lines were almost twice or more compared to many of the susceptible lines in different groups. The relative water content was almost 30% more than the susceptible genotypes. Additionally, from the rain-out experiment, we found that HKI 1532 possessed “stay green” trait, which is a desirable trait for drought tolerance. In addition to that the root length was maximum (41.8 cm) and the RWC was significantly high (65.2%) in HKI 1532 than many other susceptible genotypes. On the other hand, Clusters A-1, A-3 and cluster B each of them had two genotypes (HKI 1345 and HKI PC3, CM 144 and CM 151, and G18Seq C5 F100-1-1 and G18Seq C5 F68-2-1, respectively) and cluster C-3 had CML 69, which can serve as susceptible parents while generating mapping population. The difference between anthesis and silking was 6 days in all these susceptible genotypes except in G18Seq C5 F100-1-1, where it was 3 days. It would be now possible to map QTLs for root length, chlorophyll content, RWC, LAI and ASI using these contrasting parental lines.
Moreover, the selected parental lines were genetically distant which was confirmed by the genetic distance matrix. The genetic distance for most of the selected parental pair for mapping population development was more than 0.5 units (Table 5). The drought tolerant and susceptible genotypes from different groups were selected based on their genetic distance and morpho-physiological traits for development of new mapping population to map QTLs.
This study also identified distinct groups based on their response to drought stress and genetic relationship. Subgroups A-4, C-1 and C-4 possessed several drought tolerant lines, where as subgroups A-1, A-3 and group B had a lot of susceptible lines. These groups were genetically different from each other and having differential response to drought stress. It would be advantageous to select inbreds from the tolerant and susceptible groups, to exploit the heterosis and maximizing yield under drought stress. The lines selected for mapping population development would also serve as parental pairs for exploiting heterosis. The selected parental pairs can be tested for combining ability and for their heterotic performance.
Abbreviations
- RWC:
-
Relative water content
- ASI:
-
Anthesis-silking interval
- RL:
-
Root length
- CC:
-
Chlorophyll content
- LAI:
-
Leaf area index
- QTL:
-
Quantitative trait locus
References
Ajmone-Marsan P, Castiglioni P, Fusari F, Kuiper M, Motto M (1998) Genetic diversity and its relationship to hybrid performance in maize as revealed by RFLP and AFLP markers. Theor Appl Genet 96:219–227
Barata C, Carena MJ (2006) Classification of North Dakota maize inbred lines into heterotic groups based on molecular and testcross data. Euphytica 151:339–349
Beck DL, Vasal SK, Crossa J (1991) Heterosis and combining ability among subtropical and temperate intermediate-maturity maize germplasm. Crop Sci 31:68–73
Chin ECL, Senior ML, Shu H, Smith JSC (1996) Maize simple repetitive DNA sequences: abundance and allele variation. Genome 39:866–873
Dickson Ng’uni, Mulatu Geleta, Tomas Bryngelsson (2011) Genetic diversity in sorghum (Sorghum bicolor (L.) Moench) accessions of Zambia as revealed by simple sequence repeats (SSR) 148:52–62.
Dubreuil P, Charcosset A (1999) Relationships among maize inbred lines and populations from European and North-American origins as estimated using RFLP markers. Theor Appl Genet 99:473–480
Dubreuil P, Dufour P, Krejci E, Causse M, de Vienne D, Gallais A, Charcosset A (1996) Organization of RFLP diversity among inbred lines of maize representing the most significant heterotic groups. Crop Sci 36:790–799
Enoki H, Sato H, Koinuma K (2002) SSR analysis of genetic diversity among maize inbred lines adapted to cold regions of Japan. Theor Appl Genet 104:1270–1277
GENSTAT 14 Committee. 2011. GENSTAT 6 reference manual. Clarendon Press, Oxford, UK.
Chenyang H, Wang L, Ge H, Dong Y, Zhang X (2011) Genetic diversity and linkage disequilibrium in Chinese Bread Wheat (Triticum aestivum L.) revealed by SSR markers. PLoS ONE 6(2):e17279
Gethi JG, Labate JA, Lamkey KR, Smith ME, Kresovich S (2002) SSR variation in important US maize inbred lines. Crop Sci 42:951–957
Goodman MM, Bird RM (1977) The races of maize IV: tentative grouping of 219 Latin American races. Econ Bot 31:204–221
Heckenberger M, Melchinger AE, Ziegle JS, Joe LK, Hauser JD, Hutton M, Bohn M (2002) Variation of DNA fingerprints among accessions within maize inbred lines with regard to the identification of essentially derived varieties. I. Genetic and technical sources of variation in SSR data. Mol Breed 10:181–191
Jiang H-F, Ren X-P, Zhang X-J, Huang J-Q, Lei Y, Yan L-Y, Liao B-S, Upadhyaya HD, Holbrook CC (2010) Comparison of genetic diversity based on SSR markers between peanut mini core collections from China and ICRISAT. Acta Agron Sin 36:1084–1091
Le Clerc V, Bazante F, Baril C, Guiard J, Zhang D (2005) Assessing temporal changes in genetic diversity of maize varieties using microsatellite markers. Theor Appl Genet 110:294–302
Legesse BW, Myburg AA, Pixley KV, Botha AM (2007) Genetic diversity of African maize inbred lines revealed by SSR markers. Hereditas 144:10–7
Liu K, Muse SV (2005) PowerMarker: integrated analysis environment for genetic marker data. Bioinformatics 21:2128–2129
Liu ZW, Biyashev R, Saghai-Maroof M (1996) Development of simple sequence repeats DNA markers and their integration into a barley linkage map. Theor Appl Genet 93:869–876
Liu K, Goodman M, Muse S et al (2003) Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 165:2117–2128
Lu H, Bernardo R (2001) Molecular marker diversity among current and historical maize inbreds. Theor Appl Genet 103:613–617
Matsuoka Y, Mitchell SE, Kresovich S (2002) Microsatellites in Zea -variability, patterns of mutations and use for evolutionary studies. Theor Appl Genet 104:436–450
Melchinger AE, Gumber RK (1998) Overview of heterosis and heterotic groups in agronomic crops. In: Lamkey KR, Staub JE (eds) Concepts and breeding of heterosis in crop plants. CSSA, Madison, pp 29–44
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Perrier X, Flori A, Bonnot F (2003) Data analysis methods. In: Hamon P, Seguin M, Perrier X, Glaszmann JC (eds) Genetic diversity of cultivated tropical plants. Science Publishers Montpellier, Enfield, pp 43–76
Pinto LR, Vieira MLC, de Souza CL et al (2003) Genetic diversity assessed by microsatellites in tropical maize population submitted to high-density reciprocal recurrent selection. Euphytica 134:277–286
Reif JC, Melchinger AE, Xia XC, Warburton ML et al (2003) Use of SSRs for establishing heterotic groups in subtropical maize. Theor Appl Genet 107:947–957
Ron Parra J, Hallauer AR (1997) Utilization of exotic maize germplasm. Plant Breed Rev 14:165–187
Saker MM, Youssef SS, Abdallah NA, Bashandy HS et al (2005) Genetic analysis of some Egyptian rice genotypes using RAPD, SSR and AFLP. Afr J Biotechnol 4:882–890
Schug MD, Hutter CM, Wetterstr KA et al (1998) The mutation rate of di, tri and tetra-nucleotide repeats in Drosophila melanogaster. Mol Biol Evol 15:1751–1760
Semagn K, Bjørnstad A, Ndjiondjop MN (2006) Principles, requirements and prospects of genetic mapping in plants. African J Biotech 5:2569–2587
Senior ML, Chin ECL, Lee M et al (1996) Simple sequences repeat markers developed from maize sequences found in the GENEBANK database: map construction. Crop Sci 36:1676–1683
Senior ML, Murphy JP, Goodman MM et al (1998) Utility of SSRs for determining genetic similarities and relationships in maize using agarose gel system. Crop Sci 38:1088–1098
Sharon EM, Kresovich S, Jester CA, Hernandez CJ, Szewc-McFadden AK (1997) Application of multiplex PCR and fluorescence-based, semi-automated allele sizing technology for genotyping plant genetic resources. Crop Sci 37:617–624
Singh NN, Venkatesh S, Sekhar JC, Zaidi PH (2005) Stresses on maize in the tropics - progress and challenges. In: Stresses on maize in tropics. Directorate of Maize Research, New Delhi, India
Smart RE, Bingham GE (1974) Rapid estimates of relative water content. Plant Physiol 53:258–260
Smith JSC, Smith OS (1992) Fingerprinting crop varieties. Adv Agron 47:85–140
Smith JSC, Chin ECL, Shu H, Smith OS, Wall SJ, Senior ML, Mitchell SE, Kresovich S, Ziegle J (1997) An evaluation of the utility of SSR loci as molecular markers in maize (Zea mays L.): comparisons with data from RFLPs and pedigree. Theor Appl Genet 95:163–173
Souza SGH, Carpentieri-Pípolo V, Ruas CF, Carvalho VP et al (2008) Comparative analysis of genetic diversity among the maize inbred lines (Zea mays L.) obtained by RAPD and SSR markers. Braz Arch Biol Technol 51:183–192
Tautz D (1989) Hypervariablity of simple sequences as a general source of polymorphic DNA markers. Nucleic Acids Res 17:6463–6471
Tóth G, Gáspári Z, Jurka J (2000) Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res 10:967–981
Van Inghelandt D, Melchinger AE, Lebreton C, Stich B (2010) Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor Appl Genet 120:1289–1299
Vaz Patto MC, Satovic Z, Pego S et al (2004) Assessing the genetic diversity of Portuguese maize germplasm using microsatellite markers. Euphytica 137:63–72
Vigouroux Y, McMullen M, Hittinger CT, Houchins K, Schulz L, Kresovich S, Matsuoka Y, Doebley J (2002) Identifying genes of agronomic importance in maize by screening microsatellites for evidence of selection during domestication. Proc Natl Acad Sci 99:9650–9655
Warburton ML, Xianchun X, Crossa J et al (2002) Genetic characterization of CIMMYT inbred maize lines and open pollinated populations using large scale fingerprinting methods. Crop Sci 42:1832–1840
Warburton ML, Reif JC, Frisch M, Bohn M, Bedoya C, Xia XC, Crossa J, Franco J, Hoisington D, Pixley K, Taba S, Melchinger AE (2008) Genetic diversity in CIMMYT non-temperate maize germplasm: landraces, open pollinated varieties, and inbred lines. Crop Sci 48:617–624
Acknowledgement
The authors sincerely acknowledge the National Agricultural Innovation Project (NAIP) for their financial support to carry out this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nepolean, T., Singh, I., Hossain, F. et al. Molecular characterization and assessment of genetic diversity of inbred lines showing variability for drought tolerance in maize. J. Plant Biochem. Biotechnol. 22, 71–79 (2013). https://doi.org/10.1007/s13562-012-0112-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-012-0112-7