
Abstract
Purpose
Despite advances that have been made in systemic chemotherapy, the prognosis of advanced triple-negative breast cancer (TNBC) patients is still poor. The identification of key factors governing TNBC development is considered imperative for the development of novel effective therapeutic approaches. Previously, it has been reported that microRNA (miR)-761 may act as either a tumor suppressor or as an oncogene in different types of cancer. Here, we aimed at assessing the biological role of this miRNA in TNBC.
Methods
First, we measured the expression of miR-761 in primary breast cancer tissues and breast cancer-derived cell lines using qRT-PCR. Subsequently, over-expression and silencing experiments were performed to determine the role of miR-761 in TNBC cell proliferation, colony formation, migration and invasion in vitro. The in vivo role of miR-761 in TNBC growth and metastasis was determined in mouse models. Bioinformatics analyses, dual-luciferase reporter assays, Western blot analyses and rescue experiments were performed to identify miR-761 target gene(s).
Results
We found that miR-761 was up-regulated in primary breast cancer tissues and its derived cell lines and, particularly, in TNBC tissues and cell lines. We also found that exogenous miR-761 over-expression augmented in vitro TNBC cell proliferation, colony formation, migration and invasion, whereas miR-761 down-regulation impaired these features. In vivo, we found that miR-761 over-expression facilitated TNBC growth and lung metastasis. Mechanistically, miR-761 was found to negatively regulate the expression of tripartite motif-containing 29 (TRIM29) in TNBC cells by binding to the 3′-untranslated region of its mRNA. In conformity with these results, a significant negative correlation between miR-761 expression and TRIM29 protein expression was noted in primary TNBC tissues (r = −0.452, p = 0.0126). We also found that exogenous TRIM29 over-expression reversed the proliferative and invasive capacities of TNBC cells.
Conclusions
Our data indicate that miR-761 acts as an oncogene in TNBC. This mode of action can, at least partially, be ascribed to the down-regulation of its target TRIM29. We suggest that miR-761 may serve as a promising therapeutic target for TNBC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
World-wide, breast cancer is the most common type of cancer and a leading cause of cancer-related death among women [1]. Triple-negative breast cancer (TNBC) constitutes a heterogeneous subgroup of breast cancers, accounting for 15–20% of all cases [2–5]. TNBCs are associated with a rapid growth and early metastasis [6]. Although systemic chemotherapy is commonly used to treat advanced breast cancer [7], the prognosis of TNBC patients is still poor. The identification of key genes governing TNBC growth and metastasis is considered imperative for the development of novel effective therapeutic approaches.
microRNAs (miRNAs) constitute a group of endogenous, small, non-protein coding RNAs that are implicated in various physiological and pathological processes such as growth, differentiation, inflammation and tumorigenesis [8]. It has amply been shown that miRNAs can regulate gene expression at the post-transcriptional level by directly binding to specific sites within the 3′-untranslated regions (3′-UTR) of target mRNAs [9–11]. So far, several miRNAs have been found to be involved in the biology of TNBCs [12, 13]. It has, for example, been reported that miR-200a may suppress TNBC cell migration [12]. Another less well characterized miRNA, miR-761, has previously been linked to tumor progression, in particular in non-small cell lung cancer and miR-761 down-regulation has been found to suppress hepatocellular carcinoma growth and metastasis both in vivo and in vitro [14–16]. In ovarian carcinoma cells, however, miR-761 over-expression has been found to lead to proliferation and invasion inhibition [15]. These results suggest that miR-761 may act either as an oncogene (oncomir) or as a tumor suppressor depending on the cellular context.
Here, we aimed to assess the expression level and biological role of miR-761 in TNBC and to identify the direct target gene(s) that mediate the activity of this miRNA in TNBC.
2 Material and methods
2.1 Tissue samples
In this study we included 56 pairs of primary breast cancers and adjacent noncancerous breast tissues from 19 TNBC and 37 non-TNBC patients. The clinicopathological characteristics of the patients and tumors are listed in Table 1. Concerning age, histological grade, T- and N-stage and histological type, comparable baseline characteristics were observed between the TNBC and non-TNBC samples. Freshly obtained tissue samples were pathologically confirmed at the Department of Pathology of Zhengzhou University (Zhengzhou, China) and snap-frozen until use. None of the patients received any anticancer treatment before sample collection. Written informed consent was obtained from each participant. The study protocol was approved by the Ethics Committee of Zhengzhou University.
2.2 Cell culture
The human TNBC cell lines MDA-MB-231, MDA-MB-453, HCC1806 and MDA-MB-468, the non-TNBC cell lines MCF-7 and T-47D, and the non-tumorigenic mammary epithelial cell line MCF10A were obtained from the American Type Culture Collection (Manassas, VA, USA). The 293T cell line was obtained from the Shanghai Institutes for Biological Sciences Cell Resource Center (Shanghai, China). All the cells were cultured in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA) at 37 °C in a humidified incubator with 5% CO2.
2.3 RNA extraction and quantification
Total RNA was extracted from tissues and cells using a miRNA extraction kit (Tiangen, Beijing, China). The RNAs were reverse transcribed to cDNA using a TaqMan microRNA Reverse Transcription Kit, according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA, USA). Subsequent quantitative real-time PCR (qRT-PCR) was performed using a TaqMan Universal PCR Master Mix (Applied Biosystems). Relative miR-761 levels were normalized to U6 RNA and calculated using the 2(−ΔΔCt) method [17].
2.4 Plasmids and oligonucleotides
A fragment containing the miR-761 precursor and its flanking sequences was obtained by PCR from human genomic DNA and cloned into a pCMV vector (Origene, Nockville, MD, USA). For stable down-regulation of miR-761, a lentivral vector-based anti-miR-761 expression system (pmiRZIP-761) was purchased from System Biosciences (Mountain View, CA, USA). A tripartite motif-containing 29 (TRIM29) expression vector was generated by cloning the human TRIM29 open reading frame (Origene) into a pcDNA3.1(+) vector (Invitrogen, Carlsbad, CA, USA). 3′-UTR reporter constructs were generated by cloning the wild-type or mutated TRIM29 3′-UTR downstream of the luciferase gene in a pMIR-REPORT vector (Ambion, Carlsbad, CA, USA). In the mutated reporter construct, a putative miR-761 target in the TRIM29 3′-UTR was disrupted using a QuickChange Site-Directed Mutagenesis Kit (Stratagene, Heidelberg, Germany). miR-761 mimic, anti-miR-761 inhibitor and negative controls were purchased from the Shanghai Gene-Pharma Company (Shanghai, China).
2.5 Transfection, lentivirus production and infection
miR-761 mimic, anti-miR-761 inhibitor, and their respective controls were transfected into breast cancer cells at a final concentration of 50 nM, using Lipofectamine RNAiMax (Invitrogen). For the establishment of stable miR-761 overexpressing clones, MDA-MB-231 cells were transfected with pCMV-miR-761 or an empty vector and selected in the presence of G418 (800 μg/ml; Sigma-Aldrich) for 2 weeks. To generate stable miR-761 knockdown clones, a lentiviral expression system was employed to deliver anti-miR-761 into breast cancer cells. In brief, 293T cells were transfected with 1 μg pmiRZIP-761 and 4 μg packaging vector using Lipofectamine 2000 (Invitrogen). Next, lentivirus-containing supernatants were collected 72 h after transfection and used to infect MDA-MB-453 cells. Transduced MDA-MB-453 cells were selected using puromycin (2 μg/ml; Sigma-Aldrich) for 2 weeks to establish stable cell lines. In order to test whether TRIM29 over-expression can rescue miR-761-induced phenotypes, MDA-MB-231 cells were co-transfected with a pcDNA3.1-TRIM29 plasmid or empty vector (1 μg) together with miR-761 mimic (50 nM) and tested for gene expression, proliferation and invasion 24 h after transfection.
2.6 Cell proliferation assay
Transiently transfected cells were harvested 24 h after transfection and seeded at a density of 4 × 103 cells/well into 96-well plates and cultured for 48 or 72 h. Next, cell viabilities were determined using a CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) according to the instructions of the manufacturer.
2.7 Colony formation assay
In order to determine their colony forming capacity, cells were seeded into 6-well plates at a density of 600 cells per well. After a culture period of 14 days, the cells were fixed with methanol and stained with 0.1% crystal violet (Sigma-Aldrich). Colonies consisting of >50 cells were counted using bright field microscopy.
2.8 Transwell migration and invasion assays
The migration and invasion capacities of the cells were evaluated using Transwell chambers (Millipore, Billerica, MA, USA). For the migration assay, transfected cells were re-suspended in DMEM supplemented with 0.1% FBS and seeded into the upper Transwell chambers (24-well), and fresh medium with 10% FBS was placed in the lower chambers. For the invasion assay, transfected cells were seeded into the upper Transwell chambers pre-coated with 0.5 mg/ml Matrigel (BD Biosciences, Bedford, MA, USA). After incubation for 48 h, the insert membranes were fixed. The cells that had migrated or invaded through the chamber membrane were stained with 0.5% crystal violet and counted under an inverted microscope.
2.9 Xenograft studies
Female BALB/c nude mice (4–6 weeks old) were used for xenografting. MDA-MB-231 cells stably expressing miR-761 or containing an empty vector (1 × 106 cells per mouse) were subcutaneously injected into the flanks of mice (4 mice per group). Tumor volumes were measured every week and 4 weeks after xenografting the mice were sacrificed and the tumors were weighed.
For assessing lung metastasis, miR-761-overexpressing or control MDA-MB-231 cells (4 × 106) were injected intravenously into the tail vein. At 4 weeks after inoculation, the mice were sacrificed and the lungs were removed and subjected to pathological analysis using hematoxylin and eosin (H&E) staining. All animal experiments were performed in accordance with the protocols approved by the Institutional Review Board of Zhengzhou University.
2.10 Dual-luciferase reporter assay
293T cells were transfected with 0.2 μg of the respective 3′-UTR reporter constructs, 0.02 μg pRL-TK (Promega) and 50 nM miR-761 mimic or control miRNA. The pRL-TK vector, expressing Renilla luciferase, was used as internal control for luciferase activity. At 24 h after transfection the cells were lysed and luciferase activities were measured using a dual luciferase assay kit (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity.
2.11 Western blotting
Protein samples from primary tissues and cell lines were prepared using a radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Total proteins were resolved by sodium dodecyl-sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. These membranes were probed with a rabbit anti-TRIM29 polyclonal antibody (1:500; ab22207, Abcam, Cambridge, MA, USA) or a mouse anti-β-actin monoclonal antibody (1:3000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4 °C, followed by suitable horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma-Aldrich). Finally, protein bands were visualized using a chemiluminescent system (Cell Signaling Technology, Danvers, MA, USA) and quantified using the Quantity One software tool (Bio-Rad Laboratories, Hercules, CA, USA).
2.12 Statistical analyses
Data are presented as mean ± SEM and analyzed by Student’s t test, Mann-Whitney U test, or one-way analysis of variance (ANOVA). A Pearson correlation analysis was carried out to assess correlations between miR-761 and TRIM29 expression. A p value < 0.05 was considered statistically significant.
3 Results
3.1 miR-761 expression is up-regulated in primary TNBC tissues and TNBC-derived cell lines
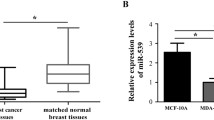
Using qRT-PCR, we found that miR-761 was up-regulated in primary breast cancer tissues compared to its corresponding normal breast tissues (p = 0.0098; Fig. 1a). Specifically, we found that miR-761 expression was significantly up-regulated in TNBC tissues compared to non-TNBC tissues (p = 0.0136; Fig. 1b). We also assessed the expression of miR-761 in a panel of breast cancer-derived cell lines. Compared to non-malignant MCF10A mammary epithelial cells, a significant (p < 0.05) increase in miR-761 expression was observed in the breast cancer-derived cells, particularly in the TNBC-derived MDA-MB-453 and MDA-MB-468 cells (Fig. 1c). These cells showed significantly (p < 0.05) higher miR-761 expression levels than the non-TNBC cells MCF-7 and T-47D. Together, these data indicate that miR-761 is up-regulated in TNBC tissues and cells.

miR-761 expression is up-regulated in in TNBC tissues and cells. a miR-761 expression in primary breast cancer tissues and its corresponding normal tissues. b miR-761 expression in TNBC and non-TNBC tissues. c miR-761 expression in breast cancer-derived cell lines. * p < 0.05 versus non-malignant MCF10A mammary epithelial cells; # p < 0.05 versus non-TNBC MCF-7 or T-47D cells
3.2 miR-761 enhances TNBC cell proliferation and colony formation
Next, we set out to investigate the role of miR-761 in TNBC cell proliferation and growth. We found that exogenous over-expression of miR-761 enhanced both the proliferation (Fig. 2a) and colony formation (Fig. 2b) capacities of MDA-MB-231 cells compared to untransfected control cells. In contrast, we found that miR-761 expression knockdown resulted in a significant decline in the proliferation (Fig. 2c) and colony formation (Fig. 2d) capacities of MDA-MB-231 cells. Very similar results were obtained with MDA-MB-453 cells (data not shown). These results suggest a growth-promoting role of miR-761 in TNBC cells.

miR-761 promotes TNBC cell proliferation and colony formation. a cell proliferation and b colony formation of MDA-MB-231 cells transfected with a control miRNA or miR-761. c and d proliferation and colony formation of MDA-MB-231 cells transfected with a control miRNA or anti-miR-761. Bar graphs represent results from three independent experiments. * p < 0.05 versus control
3.3 miR-761 enhances TNBC cell migration and invasion
We also assessed the ability of miR-761 to regulate TNBC cell migration and invasion. By using Transwell migration and invasion assays, we found that exogenous miR-761 expression significantly (p < 0.05) increased the migration and invasion capacities of MDA-MB-453 cells, whereas miR-761 expression knockdown led to a significant inhibition of the migration and invasion capacities of these cells (Fig. 3a and b). Very similar results were obtained with MDA-MB-231 cells (Fig. 3c and d). These results suggest that miR-761 exerts a pro-invasive activity in TNBC cells.

miR-761 enhances TNBC cell migration and invasion. a and b MDA-MB-453 cells and c and d MDA-MB-231 cells were transfected with miR-761, anti-miR-761 or its corresponding controls and subjected to Transwell migration (a and c) and invasion (b and d) assays. Bar graphs represent quantitative data from three independent experiments. * p < 0.05 versus control
3.4 miR-761 over-expression enhances TNBC growth and metastasis in vivo
Next, we explored the effect of miR-761 over-expression on TNBC growth and metastasis in vivo. To this end, MDA-MB-231 cells stably transfected with a miR-761 expression vector or an empty vector were injected subcutaneously into the flanks of nude mice after which tumor growth was monitored. We found that the miR-761 over-expressing MDA-MB-231 xenografts yielded significantly (p < 0.05) higher tumor volumes than the control (empty vector) xenografts (Fig. 4a). The average tumor weight in the miR-761 over-expression group was found to be increased by 85% 4 weeks after inoculation compared to the control group (p = 0.0241; Fig. 4b). Subsequent Western blot analysis revealed that the miR-761 over-expression group exhibited significantly lower TRIM29 levels (see below) than the control group (p = 0.0163; Fig. 4c).

miR-761 over-expression enhances TNBC cell growth and metastasis in vivo. MDA-MB-231 cells stably transfected with a miR-761 expression plasmid or an empty vector were injected subcutaneously into nude mice. a Xenograft tumor growth curves based on tumor volumes. b Tumors removed, photographed and weighed 4 weeks after inoculation. c Western blot analysis of TRIM29 in xenograft tumors as shown in (b). Lanes 1–4 represent 4 different tumors. miR-761 expression or empty vector transfected MDA-MB-231 cells were injected into nude mice via the tail vein and lung metastatic lesions were assessed 5 weeks after inoculation. d Representative tissue sections of lungs with metastatic lesions (arrows). Scale bar: 100 μm. e Significant increase in the number of metastatic lesions in the miR-761 over-expression group compared to the control group. * p < 0.05 versus control
We also injected miR-761 over-expression and empty vector-transfected MDA-MB-231 cells into the tail vein of nude mice and assessed the occurrence of metastatic lesions in the lungs 5 weeks after inoculation. We found that the number of lung metastatic nodules was significantly increased in the miR-761 over-expression group compared to the control empty vector group (p = 0.0019) (Fig. 4d and e). These results indicate that miR-761 enhances both the tumorigenicity and metastatic spread of TNBC cells in vivo.
3.5 miR-761 directly targets the 3′-UTR of TRIM29
In order to uncover the molecular mechanism underlying the tumor-promoting activity of miR-761 in TNBC cells, we sought to identify its direct target genes. Bioinformatic analysis using the miRDB software tool (http://mirdb.org/miRDB/) revealed that TRIM29 may serve as a potential target of miR-761, since the 3′-UTR of its mRNA contains a putative target site for miR-761 (Fig. 5a). Subsequently, we noticed that in TNBC tissues, there is a significant negative correlation between miR-761 and TRIM29 expression (r = −0.452, P = 0.0126; Fig. 5b). In order to confirm that miR-761 can negatively regulate TRIM29 expression by binding to its 3′-UTR, we cloned both a wild-type and mutated TRIM29 3′-UTR downstream to the luciferase reporter gene and performed luciferase reporter assays. By doing so, we found that exogenous miR-761 expression significantly (p < 0.05) decreased the activity of the reporter harboring the wild-type TRIM29 3′-UTR (Fig. 5c), whereas the activity of the reporter harboring the mutated TRIM29 3′-UTR was not altered by exogenous miR-761 expression. Consistent with the reporter assay, we found that the endogenous TRIM29 expression in MDA-MB-231 cells was significantly reduced after miR-761 mimic delivery (Fig. 5d), whereas transfection with anti-miR-761 significantly increased the expression of TRIM29. Taken together, we conclude that miR-761 has an adverse impact on the expression of TRIM29 in TNBC cells.

miR-761 directly targets the 3′-UTR of TRIM29. a Bioinformatic analysis revealing a putative miR-761 binding site within the 3′-UTR of TRIM29. Disruption of the predicted miR-761 binding site was performed by site-directed mutagenesis (mutant). b Spearman correlation analysis of miR-761 and TRIM29 expression levels in 19 primary TNBC specimens. c 293T cells were co-transfected with reporter constructs and a miR-761 mimic or control miRNA. Firefly luciferase activity was normalized to that of Renilla luciferase. d Western blot analysis of TRIM29 in MDA-MB-231 cells transfected with miR-761, anti-miR-761 and its corresponding controls. Bar graphs represent quantitative data from three independent experiments. * p < 0.05 versus control
3.6 TRIM29 over-expression attenuates miR-761-induced proliferation and invasion of TNBC cells
Based on previous reports indicating that TRIM29 may act as a tumor suppressor in breast cancer [18, 19], we asked whether TRIM29 down-regulation may account for the miR-761-mediated aggressive phenotype of TNBC cells. To address this issue, we performed rescue experiments with a TRIM29 variant lacking the 3′-UTR. We found that when this plasmid was co-transfected into MDA-MB-231 cells together with miR-761 mimic, the miR-761-mediated down-regulation of TRIM29 was fully abolished (Fig. 6a). Most interestingly, we found that TRIM29 over-expression impaired the proliferation (Fig. 6b) and invasion (Fig. 6c) of MDA-MB-231 cells induced by miR-761 mimic. These data suggest that TRIM29 down-regulation may underlie the tumor-promoting effect of miR-761 in TNBC cells.

Exogenous TRIM29 over-expression attenuates miR-761-induced TNBC growth and invasion. a Western blot analysis of TRIM29 in MDA-MB-231 cells transfected with a miR-761 mimic, or together with a TRIM29 expression plasmid or an empty vector. b MDA-MB-231 cells transfected with the indicated constructs were cultured for 48 or 72 h and assessed for their proliferative capacity. c MDA-MB-231 cells transfected with the indicated constructs were subjected to Transwell invasion assays. Bar graphs represent quantitative data from three independent experiments. * p < 0.05 versus control; # p < 0.05 versus miR-761 transfected cells
4 Discussion
Previously, it has been reported that a vast number of miRNAs is differentially expressed between breast cancer tissues and normal breast tissues [20–22]. Only a minority of them has, however, been functionally characterized. Here, we show that miR-761 expression is significantly up-regulated in breast cancer tissues and to a greater extent TNBC tissues. This expression trend could be confirmed in breast cancer-derived cell lines. Up-regulation of miR-761 has also been observed in several other cancer types, including non-small cell lung cancer [14] and hepatocellular carcinoma [16]. To validate the biological relevance of miR-761 up-regulation in TNBC cells, we performed expression modulation studies. We found that exogenous miR-761 expression enhanced the proliferation, colony formation, migration and invasion capacities of MDA-MB-231 cells, whereas miR-761 expression knockdown significantly suppressed these capacities. Subsequent in vivo studies showed that miR-761 over-expression significantly promotes the growth and metastasis of MDA-MB-231 xenograft tumors, thereby confirming the oncogenic role of miR-761 in TNBC.
A previous study has shown that miR-761 can augment hepatocellular carcinoma cell proliferation and invasion by targeting mitofusin-2 [16]. Although it has been found that mitofusin-2 can inhibit non-TNBC cell proliferation and invasion [23], we failed to find any significant association between miR-761 expression and mitofusin-2 expression in the breast cancer samples that we tested (data not shown). Therefore, this miRNA may exert its effects on breast cancer cells via a mitofusin-2-independent mechanism.
We identified TRIM29 as a potential target of miR-761 using bioinformatics and luciferase reporter assays. This notion was substantiated by our finding that exogenous miR-761 expression resulted in a significant reduction in endogenous TRIM29 expression, while miR-761 silencing significantly promoted TRIM29 expression in MDA-MB-231 cells. These results strongly indicate that TRIM29 acts as a genuine target of miR-761. TRIM29 has been found to exhibit tumor-promoting activities in several types of cancer, such as pancreatic cancer [24], nasopharyngeal carcinoma [25] and gastric cancer [26]. Several lines of evidence indicate that there are links between TRIM29 expression and miRNA regulation [26, 27]. In gastric cancer cells, for example, TRIM29 has been found to act as a functional target of miR-185 [26], whereas TRIM29 over-expression has been found to promote the invasive growth in bladder cancer cells by suppressing miR-29 [27]. Compelling evidence indicates that TRIM29 plays a suppressive role in breast cancer [18, 19]. It has e.g. been reported that TRIM29 silencing causes malignant transformation of MCF10A human epithelial breast cancer cells, facilitating their proliferation, anchorage-independent growth and invasion [19]. Another study has reported that expression knockdown of TRIM29 promotes breast cancer cell migration and invasion through induction of TWIST expression and epithelial-mesenchymal transition [18]. These studies suggest that TRIM29 repression may account for the miR-761-mediated induction of aggressive phenotypes in TNBC cells. In support of this hypothesis, we found that TRIM29 over-expression impaired the proliferation and invasion of MDA-MB-231 cells induced by miR-761. Subsequent xenograft studies confirmed that miR-761 over-expression, and its concomitant down-regulation of TRIM29, impaired the growth and metastasis of MDA-MB-231 tumors. Additional primary clinical TNBC data also revealed a significant negative correlation between miR-761 expression and TRIM29 expression. Taken together, these results suggest that miR-761 exerts its oncogenic activity in TNBC cells through targeting TRIM29.
It needs to be mentioned, however, that miR-761 may also target other tumor suppressors such as ING4 and TIMP2 [14], and it has been found that TIMP2 is capable of inhibiting breast cancer cell migration and invasion [28] and that adenoviral delivery of ING4 can lead to the suppression of breast cancer growth in vitro and in vivo [29]. Therefore, additional studies are required to test whether the oncogenic activity of miR-761 should be ascribed to the modulation of multiple tumor suppressors.
In summary, we provide evidence that miR-761 can promote the proliferation, migration and invasion of TNBC cells in vitro and accelerate the growth and metastasis of TNBC cells in vivo. Mechanistically, we found that TRIM29 down-regulation may account, at least partially, for the miR-761-induced aggressive phenotype of TNBC cells. Our data suggest that miR-761 may serve as a potential therapeutic target for TNBC.
References
L.A. Torre, F. Bray, R.L. Siegel, J. Ferlay, J. Lortet-Tieulent, A. Jemal, Global cancer statistics, 2012. CA Cancer J. Clin. 65, 87–108 (2015)
G. Palma, G. Frasci, A. Chirico, E. Esposito, C. Siani, C. Saturnino, C. Arra, G. Ciliberto, A. Giordano, M. D’Aiuto, Triple negative breast cancer: looking for the missing link between biology and treatments. Oncotarget 6, 26560–26574 (2015)
I. Fkih M’hamed, M. Privat, F. Ponelle, F. Penault-Llorca, A. Kenani, Y.J. Bignon, Identification of miR-10b, miR-26a, miR-146a and miR-153 as potential triple-negative breast cancer biomarkers. Cell. Oncol. 38, 433–442 (2015)
C.B. Moelans, E.J. Vlug, C. Ercan, P. Bult, H. Buerger, G. Cserni, P.J. van Diest, P.W. Derksen, Methylation biomarkers for pleomorphic lobular breast cancer - a short report. Cell. Oncol. 38, 397–405 (2015)
E. Robles-Escajeda, U. Das, N.M. Ortega, K. Parra, G. Francia, J.R. Dimmock, A. Varela-Ramirez, R.J. Aguilera, A novel curcumin-like dienone induces apoptosis in triple-negative breast cancer cells. Cell. Oncol. 39, 265–277 (2016)
J. Hugh, J. Hanson, M.C. Cheang, T.O. Nielsen, C.M. Perou, C. Dumontet, J. Reed, M. Krajewska, I. Treilleux, M. Rupin, E. Magherini, J. Mackey, M. Martin, C. Vogel, Breast cancer subtypes and response to docetaxel in node-positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. J. Clin. Oncol. 27, 1168–1176 (2009)
J. Collignon, L. Lousberg, H. Schroeder, G. Jerusalem, Triple-negative breast cancer: treatment challenges and solutions. Breast Cancer 8, 93–107 (2016)
Q. Huang, B. Xiao, X. Ma, M. Qu, Y. Li, P. Nagarkatti, M. Nagarkatti, J. Zhou, MicroRNAs associated with the pathogenesis of multiple sclerosis. J. Neuroimmunol. 295-296, 148–161 (2016)
M. Tomasetti, M. Amati, L. Santarelli, J. Neuzil, microRNA in metabolic re-programming and their role in tumorigenesis. Int. J. Mol. Sci. 17, 754 (2016)
V. Taucher, H. Mangge, J. Haybaeck, Non-coding RNAs in pancreatic cancer: challenges and opportunities for clinical application. Cell. Oncol. 39, 295–318 (2016)
A. Ferraro, Altered primary chromatin structures and their implications in cancer development. Cell. Oncol. 39, 195–210 (2016)
E. Tsouko, J. Wang, D.E. Frigo, E. Aydoğdu, C. Williams, miR-200a inhibits migration of triple-negative breast cancer cells through direct repression of the EPHA2 oncogene. Carcinogenesis 36, 1051–1060 (2015)
X. Sui, X. Wang, W. Han, D. Li, Y. Xu, F. Lou, J. Zhou, X. Gu, J. Zhu, C. Zhang, H. Pan, MicroRNAs-mediated cell fate in triple negative breast cancers. Cancer Lett. 361, 8–12 (2015)
A. Yan, C. Yang, Z. Chen, C. Li, L. Cai, MiR-761 promotes progression and metastasis of non-small cell lung cancer by targeting ING4 and TIMP2. Cell. Physiol. Biochem. 37, 55–66 (2015)
C. Shi, Z. Zhang, miR-761 inhibits tumor progression by targeting MSI1 in ovarian carcinoma. Tumour Biol. 37, 5437–5443 (2016)
X. Zhou, L. Zhang, B. Zheng, Y. Yan, Y. Zhang, H. Xie, L. Zhou, S. Zheng, W. Wang, MicroRNA-761 is upregulated in hepatocellular carcinoma and regulates tumorigenesis by targeting mitofusin-2. Cancer Sci. 107, 424–432 (2016)
K.J. Livak, T.D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C (T)) method. Methods 25, 402–408 (2001)
L. Ai, W.J. Kim, M. Alpay, M. Tang, C.E. Pardo, S. Hatakeyama, W.S. May, M.P. Kladde, C.D. Heldermon, E.M. Siegel, K.D. Brown, TRIM29 suppresses TWIST1 and invasive breast cancer behavior. Cancer Res. 74, 4875–4887 (2014)
J. Liu, B. Welm, K.M. Boucher, M.T. Ebbert, P.S. Bernard, TRIM29 functions as a tumor suppressor in nontumorigenic breast cells and invasive ER+ breast cancer. Am. J. Pathol. 180, 839–847 (2012)
P. Krishnan, S. Ghosh, B. Wang, D. Li, A. Narasimhan, R. Berendt, K. Graham, J.R. Mackey, O. Kovalchuk, S. Damaraju, Next generation sequencing profiling identifies miR-574-3p and miR-660-5p as potential novel prognostic markers for breast cancer. BMC Genomics 16, 735 (2015)
E. van Schooneveld, H. Wildiers, I. Vergote, P.B. Vermeulen, L.Y. Dirix, S.J. Van Laere, Dysregulation of microRNAs in breast cancer and their potential role as prognostic and predictive biomarkers in patient management. Breast Cancer Res. 17, 21 (2015)
R. Maqbool, R. Rashid, R. Ismail, S. Niaz, N.A. Chowdri, M.U. Hussain, The carboxy-terminal domain of connexin 43 (CT-Cx43) modulates the expression of p53 by altering miR-125b expression in low-grade human breast cancers. Cell. Oncol. 38, 443–451 (2015)
L. Ma, Y. Liu, C. Geng, X. Qi, J. Jiang, Estrogen receptor β inhibits estradiol-induced proliferation and migration of MCF-7 cells through regulation of mitofusin 2. Int. J. Oncol. 42, 1993–2000 (2013)
L. Wang, D.G. Heidt, C.J. Lee, H. Yang, C.D. Logsdon, L. Zhang, E.R. Fearon, M. Ljungman, D.M. Simeone, Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and beta-catenin stabilization. Cancer Cell 15, 207–219 (2009)
X.M. Zhou, R. Sun, D.H. Luo, J. Sun, M.Y. Zhang, M.H. Wang, Y. Yang, H.Y. Wang, S.J. Mai, Upregulated TRIM29 promotes proliferation and metastasis of nasopharyngeal carcinoma via PTEN/AKT/mTOR signal pathway. Oncotarget 7, 13634–13650 (2016)
F. Qiu, J.P. Xiong, J. Deng, X.J. Xiang, TRIM29 functions as an oncogene in gastric cancer and is regulated by miR-185. Int. J. Clin. Exp. Pathol. 8, 5053–5061 (2015)
P.L. Palmbos, L. Wang, H. Yang, Y. Wang, J. Leflein, M.L. Ahmet, J.E. Wilkinson, C. Kumar-Sinha, G.M. Ney, S.A. Tomlins, S. Daignault, L.P. Kunju, X.R. Wu, Y. Lotan, M.E. Liebert, M.E. Ljungman, D.M. Simeone, ATDC/TRIM29 drives invasive bladder cancer formation through miRNA-mediated and epigenetic mechanisms. Cancer Res. 75, 5155–5166 (2015)
M.R. Clarke, F.M. Imhoff, S.K. Baird, Mesenchymal stem cells inhibit breast cancer cell migration and invasion through secretion of tissue inhibitor of metalloproteinase-1 and -2. Mol. Carcinog. 54, 1214–1219 (2015)
Z. Li, Y. Xie, W. Sheng, J. Miao, J. Xiang, J. Yang, Tumor-suppressive effect of adenovirus-mediated inhibitor of growth 4 gene transfer in breast carcinoma cells in vitro and in vivo. Cancer Biother. Radiopharm. 25, 427–437 (2010)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Rights and permissions
About this article

Cite this article
Guo, GC., Wang, JX., Han, ML. et al. microRNA-761 induces aggressive phenotypes in triple-negative breast cancer cells by repressing TRIM29 expression. Cell Oncol. 40, 157–166 (2017). https://doi.org/10.1007/s13402-016-0312-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-016-0312-6