
Abstract
Objective
To determine if molecular and immunohistochemical (IHC) features of the HRPT2/CDC73 gene and its product, parafibromin, predict the natural history of parathyroid malignancy, particularly atypical adenoma, as seen in a single-centre patient cohort.
Methods
Matched tumor and non-tumor tissues were obtained from 46 patients with parathyroid carcinoma (CA) (n = 15), atypical adenoma (AA) (n = 14) and typical adenoma (TA) (n = 17), as defined by standardized histopathological criteria. Exons and exon-intron boundaries of the CDC73 gene were sequenced to identify germline or somatic mutations. IHC staining for parafibromin was performed and scored as positive if nuclear staining was at least partially IHC-positive.
Results
Mutations of CDC73 were observed in 9/15 (60 %) CA, 2/14 (14 %) AA, and 1/17 (6 %) TA tumors. A recurrent two basepair mutation in exon 7 -- c.679_680delAG -- accounted for half of all identified mutations. Absence of parafibromin nuclear staining was noted in 8/12 (67 %) CA, 2/13 (15 %) AA, and 3/17 (18 %) TA tumors. Median follow up times were 88 months for CA, 76 months for AA, and 104 months for TA patients. One patient, a member of a previously reported multiplex family with a germline CDC73 mutation was found to have a second adenoma after removal of an atypical adenoma.
Conclusions
Molecular screening and IHC are both useful tools in the differential diagnosis of parathyroid tumors, but both have limited sensitivity and specificity. CDC73 mutations and negative immunostaining were common in atypical adenomas, but no local recurrence was observed in any case with successful surgical removal after follow-up periods of 27 to 210 months.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Primary hyperparathyroidism (PHPT) is caused by parathyroid adenoma in 85 %, hyperplasia in 15 %, and cancer in less than 1 % of cases [1, 2]. The biological behaviours of carcinoma and adenoma are clearly different but the timely pathological diagnosis of parathyroid carcinoma can be difficult. Indeed, malignancy is commonly diagnosed only after local recurrence or detection of distant metastases [3, 4].
The diagnostic criteria for parathyroid carcinoma (CA) were defined in 1973 [5], but have been recently revised to include a new class of tumour [6]. Parathyroid tumors lacking unequivocal evidence of invasion, but showing features suspicious of malignancy (i.e., fibrous bands, questionable capsular invasion, increased mitotic figures, and adherence to surrounding tissues) are now defined as ‘atypical adenomas’ [7]. Unlike parathyroid carcinoma, the natural history of the atypical adenoma (AA) is not well established, even though the recommendation is that such patients should be closely followed [7]. In order to follow these patients, improved diagnostic and predictive tools would be helpful.
Parathyroid carcinoma is a feature of the hyperparathyroidism-jaw tumor (HPT-JT) syndrome due to HRPT2/CDC73 mutation [8, 9], and is implicated in sporadic carcinoma as well [10]. Identification of CDC73 mutations or loss of expression of the CDC73 gene product, parafibromin, may both be useful markers of malignancy in parathyroid tumours [3, 11, 12].
The prevalence of CDC73 gene mutations has been reported to be 70 % of parathyroid carcinomas [3, 10, 13, 14], but only 1–2 % of typical adenomas (TA) [8, 13, 15, 16]. About 20 % of atypical adenoma (AA) cases have mutations [11, 12].
Loss of parafibromin nuclear expression, as assessed by immunohistochemical (IHC)-negativity, has been reported to vary from 68 to 100 % for parathyroid carcinoma [11, 12, 17, 18]. Different scoring systems used may no doubt contribute to variability in frequency among the various studies [7, 11, 12, 17–19]. While partially reduced expression of parafibromin has been described in 16 of 46 atypical adenomas [11, 12, 17–21], only 4 of 238 typical adenomas appear to be similarly affected [12, 17, 18, 20–22].
Strong concordance between CDC73 mutation identification and IHC-negativity for nuclear parafibromin expression has been considered evidence in support of the suggestion that IHC can be the primary means of evaluating malignancy [12, 14, 17]. Immunohistochemistry is less expensive and time-consuming than genetic screening [12], and is theoretically more likely to pick up deleterious variants leading to complete loss of parafibromin expression. However, others have more recently reported a lower success rate for ‘IHC-negativity’ in malignancy [7, 20, 21]. Some studies have evaluated molecular screening and parafibromin expression simultaneously [11, 12, 22, 23] but data on atypical adenomas are sparse, despite the fact that this is the group for which optimal assessment might be most clinically beneficial [11, 12].
We therefore undertook to identify CDC73 mutations along with IHC-assessment of nuclear parafibromin expression, in a large, single-centre cohort of patients with histopathologically proven parathyroid tumors followed for up to 20 years.
2 Subjects and methods
2.1 Study inclusion criteria
All patients admitted to the Endocrinology Unit of the “Casa Sollievo della Sofferenza” Hospital in San Giovanni Rotondo (Foggia, Italy) with a working diagnosis of primary hyperparathyroidism were eligible for inclusion. All patients whose pathological diagnosis was parathyroid carcinoma or atypical adenoma were included, while 15 typical parathyroid adenomas were randomly selected from a large (n > 150) archival pool. Two subjects with a family history of typical parathyroid adenoma, but without mutations in calcium-sensing receptor (CASR) or multiple endocrine neoplasia type I (MEN1) genes [24] were included in the latter group. First contact with the patients occurred between calendar years 1990 and 2007 and outpatient clinic follow-up extended to 2010 (Table 1).
Subject AA7 and his family have been reported previously [25, 26]. In the other probands bearing a germline mutation, specific mutation testing was offered to all available first-degree relatives, and performed on consenting subjects (Table 2).
2.2 Clinical follow-up and surveillance
All patients with parathyroid carcinoma and atypical adenoma were followed up with yearly measurement of serum calcium, creatinine and PTH plus ultrasonography of neck in order to detect the presence of early recurrence of the disease. In probands and their relatives bearing a germline mutation of the CDC73 gene, our protocol includes annual clinical examination, biochemical profile (i.e., serum ionized calcium, creatinine, PTH) and imaging (i.e., neck, total abdominal ultrasonography) with jaw pantomograms every 5 years.
2.3 Histopathologic diagnosis
For histopathological analysis at diagnosis, tissue samples were excised and fixed in 10 % buffered formalin followed by conventional processing and paraffin embedding. Four micron thick sections were cut and stained with haematoxylin and eosin for standard histopathological assessment. Parathyroid tumors were carefully re-evaluated and classified according to current WHO guidelines [6, 27]. Where discordances in IHC-negativity and molecular studies arose subsequently, the histopathological classification was independently re-evaluated in a blinded fashion. However, no discrepancies in routine histopathological classification were found.
All patients, or immediate relatives if the subject was deceased, gave informed consent. The study protocol was approved by the ethics committee of the hospital.
2.4 Molecular studies
For all subjects, whole blood and matching tumor tissues available in paraffin blocks were processed for the analysis of germline and somatic mutations, respectively. In the case of a deceased individual, non-tumor tissue was collected. DNA from peripheral leukocytes was isolated by phenol-chloroform extraction, and DNA from tissues was extracted as described previously [28].
Molecular screening of the entire CDC73 coding sequence (17 exons including exon-intron boundaries) (Fig. 1) was performed on germline DNA, and somatic DNA from all tumor samples. Amplifications [8], were carried out in a 25 μL reaction volume containing 2.5 μL 10X PCR Buffer (Eppendorf), 0.25 nM dNTPs, 20 pmol of each primer, 1 U HotMaster Taq (Eppendorf) and 100 ng of DNA. Cycling conditions consisted of initial 5 min denaturation step at 95 °C, followed by 35 cycles of 95 °C for 30 s annealing for 30 s, and extension at 72 °C for 30 s, with final extension at 72 °C for 7 min. PCR products were visualised by ethidium bromide staining on 2 % agarose gels. PCR products were purified using the GFX™ PCR and Band Purification Kit (GE Healthcare, Buckinghamshire UK) and then sequenced using the BigDye Terminator Cycle Sequencing Kit v. 1.1 (Applied Biosystems, Foster City CA). Sequencing reactions were loaded on an ABI 3100 capillary sequencer (Applied Biosystems, Foster City CA) and analysed using the Sequencing Analysis software v.3.7.

Schema showing the numbered CDC73 exons and the specific mutations identified in the present study
PCR controls included reactions without DNA. Mutations were confirmed by sequencing in both directions with forward and reverse primers on the original amplicon, and on another PCR performed on DNA re-extracted from the same tumor. Germline mutations were confirmed on the corresponding somatic tissue sample(s).
2.5 Parafibromin immunohistochemistry (IHC)
Heat-induced antigenic retrieval for detection of parafibromin was performed by placing deparaffinized 4 μm-thick sections in 10 mM citrate buffer (pH 6.0) and incubating in a 360 W microwave oven for 3 cycles of 15 min each. Tissue sections were then incubated with a primary monoclonal antibody to parafibromin (1:200 dilution; clone sc-33638 from Santa Cruz Biotechnology, Santa Cruz, CA), raised against a peptide comprising amino acids 87 to 100 in the molecule. Immunostaining was performed using an Envision™ Kit on a Dako automatic stainer (Dako, CA, USA), according to the manufacturer’s instructions. Adjacent endothelial cells served as the internal positive controls, while omission of the primary antibody served as a negative control. Slides were lightly counterstained with haematoxylin, dehydrated, and mounted. The stained sections from all cases were examined by one of us (MB), and the absence or presence of nuclear immunoreactivity was recorded as IHC-negative or IHC-positive, respectively (Table 1). At variance with some studies [11, 17, 19], but concordant with others [14, 18, 21], we considered samples IHC-negative only if they did not reveal any nuclear staining at all.
3 Results
3.1 Clinical characteristics
Our cohort consisted of 46 subjects, all of Italian heritage (Table 1). There were more women (n = 30) than men (n = 16), but the difference did not reach significance and there was no difference in their median age. The proportion of women was higher in the TA (13 of 17) and CA (10 of 15) groups, compared to the AA group (7 of 14) but the differences were not significant.
The median age at the time of operation was 54 years (range: 22 to 86). When analyzed by group, median age at first operation was 47 years for CA, 54 years for AA, and 58 years for TA subjects (p = NS). Preoperative serum total calcium (all-group median 11.7 mg/dL) was significantly higher in CA [12.9 mg/dl (range 10.6–20.2)] and AA [11.8 mg/dL (range 11.1–15.8)] groups compared to the TA [11.0 mg/dL (range 10.1–12.8)] group (ANOVA with Bonferroni post hoc analysis, p < 0.05). Preoperative PTH level (all-groups median 258 pg/mL) was also significantly higher in CA [367 pg/mL (range 60–940)] and AA [288 pg/mL (range 108–1,633)] groups vs the TA [156 pg/mL (range 55–423)] group (p < 0.05). Tumor size (all-groups median 2.3 cm) was significantly higher in the CA [3 cm (range 1.3–6 cm)] than in the TA [2.0 (range 0.4–5 cm)] group (p < 0.05). Median tumor size for the AA group was 2.3 cm (range 1.5–4 cm).
3.2 Clinical follow-up
Overall, the follow-up interval ranged from 2 months (0.2 years) to 245 months (20.4 years) with a median of 99 months (8.3 years). Four patients were lost to follow up—one with carcinoma at 88 months, two with atypical adenoma at 27 and 208 months and one with typical adenoma at 56 months.
Of the CA group, three (20 %) died of their disease at 2, 43, and 86 months after diagnosis. The rest of the group (n = 12) were monitored annually for a median interval of 99 months (range 54–245), but only one subject (CA2) presented with local recurrence and brain metastasis 219 months after diagnosis. She was still well at 245 months after radiotherapy. The AA group of 14 subjects was followed up for a median interval of 76 months. The patient (AA7) belonging to a 4-generation family with multiple affected members carrying a germline truncating mutation, reported by Guarnieri et al. 2006 [25], had a recurrence after 144 months. The 17 subjects from the TA group did not show any biochemical or ultrasonographic recurrence after a median follow-up of 104 months.
3.3 Molecular findings
In the CA group, 10 CDC73 mutations were found in 9 out of 15 subjects (60 %); three of these were germline (Table 1). A smaller proportion of the AA group (2/14 or 14 %) were mutation positive (2 germline mutations). In the TA group the number of mutation-positive subjects was still smaller (1/17 or 6 %) and there were no germline mutations. The trend across groups was statistically significant (p = 0.007, Fisher exact test).
Sites of mutation are shown in Fig. 1. In our cohort, the recurrent c.679_680delAG dinucleotide deletion accounted for 2 of the 5 germline and 5 of 9 somatic mutations (Table 2). This frequency (7/14 or 50 %) is higher (p < .001, Fisher exact test) than suggested by Masi et al. 2008 [29] in their most recent review [cf. 7/93 (8 %) independent mutations], but consistent with the view that it is a hot spot. The other seven exonic mutations were observed only once in our cohort but, of the seven, the three that were germline in our series have been reported before as germline mutations by others (Table 2). One of the three is a nonsense mutation in exon 5, while the other two are frameshift mutations in exon 7, consistent with the excess of similar germline mutations in that region. Two of the four novel exonic mutations, all somatic, were frameshift, but the other two are relatively non-conservative missense changes (c.13C>T or p.L5F, c.231C>G or p.R77P). There was no indication that missense and nonsense mutations were less common in the CA group (2 of 10 mutations) than in the other two groups (1 of 4 mutations). Both missense mutations were predicted to be damaging according to the majority (2 of 3) of the bioinformatic predictive programs used for in silico evaluation (Polyphen; L5F, benign; R77P, possibly damaging: SIFT; L5F, not tolerated; R77P, tolerated: SNPs3D; L5F, deleterious; R77P, deleterious).
Of the 14 CDC73 mutations, 7 were somatic only. Overall, somatic mutations were more frequent in the CA group (7 of 15 subjects) than in AA (1 in 14 subjects) or TA groups (1 in 17 subjects). Double hits (2 somatic or somatic+germline), consistent with the Knudson two-hit hypothesis, were found in one CA and one AA subject. In a parallel study, allelic imbalance (loss of heterozygosity = LOH) [30] was evaluated for its contribution to genetic risk (see Supplementary Material).
First-degree relatives with positive carrier status were identified in families of three probands with germline mutations (Table 2). One family has been reported in detail [25]. In the other two, all seven healthy carriers have been followed for a total of 15 patient years without identifying any evidence of disease.
3.4 Immunohistochemistry
Immunostaining of parafibromin showed loss of nuclear immunoreactivity (IHC-negativity) in 8 out of 12 CA, 2 out of 13 AA, and 3 out of 17 TA tissue samples (Table 1). Examples of tumors that are either parafibromin IHC-positive or IHC-negative, respectively, are shown for carcinomas (Fig. 2), atypical adenomas (Fig. 3) and typical adenomas (Fig. 4). The prevalence of IHC-negativity is higher in our sample of typical sporadic adenomas than reported by others [12, 17–22] despite a more conservative definition of IHC loss. Interestingly, in one of the two TA subjects without any nuclear staining for parafibromin (Fig. 4c and d) and without a positive family history (TA#8), there was a double hit, consisting of the recurrent somatic mutation, c.679_680delAG, and LOH (See Suppl Fig. 1). In the other subject (TA#2), LOH was also observed and review of the slides showed small cysts in the excised gland.

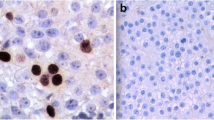
Histochemistry in carcinomas. a, c HE stain: b, d parafibromin IHC. a, b IHC-positive carcinoma from #CA9; a tumor shows a diffuse proliferation pattern with invasion of adjacent thyroid tissue (arrows) (magnification × 100; bar 52 μm); b nuclei of tumor cells along with endothelial cells and fibroblasts (arrows) are positive for parafibromin staining (magnification × 100; bar 52 μm). c, d IHC-negative carcinoma from #CA11; c diffuse follicular proliferation pattern (magnification × 100; bar 52 μm); d tumor cell nuclei are immunonegative for parafibromin while endothelial cell and fibroblast nuclei (arrows) are immunopositive. Areas of necrosis (arrowhead) are also seen (magnification × 100; 52 μm)

Histochemistry in atypical adenomas. a, c HE stain: b, d parafibromin IHC. a, b IHC-positive atypical adenoma from #AA2; a tumor cells show trabecular proliferation pattern, and apparent capsular invasion (arrows) (magnification × 200; bar 20 μm); b nuclei of tumor cells as well as endothelial cells and fibroblasts (arrows) are uniformly and strongly immunopositive (magnification × 200 bar 20 μm). c, d IH-negative atypical adenoma from #AA14; c follicular proliferation pattern (magnification × 200; bar 20 μm); d nuclei of tumor cells are immunonegative, while nuclei of fibroblasts and endothelial cells (arrows) are immunopositive (magnification × 200; bar 20 μm)

Histochemistry in typical adenomas. a, c HE stain: b, d parafibromin IHC. a, b IHC-positive typical adenoma from #TA11; a diffuse proliferation pattern and small cysts (arrows) (magnification × 200; bar 20 μm); b nuclei of tumor cells along with endothelial cells and fibroblasts (arrows) are diffusely and strongly immunopositive (magnification × 400; bar 13 μm). c, d IHC-negative typical adenoma from #TA8; c diffuse proliferation pattern (magnification × 200; bar 20 μm); d absence of immunostaining for parafibromin in tumor cells but nuclei of fibroblasts and endothelial cells (arrows) are immunopositive (magnification × 200; bar 20 μm)
3.5 Sensitivity and specificity of molecular and IHC results
Taking the clinical diagnosis as the gold standard, CDC73 mutations had diagnostic sensitivity and specificity of 60 % and 94 %, respectively. Positive and negative predictive values were 90 % and 73 %, respectively, for differentiating carcinoma from typical adenoma. With immunostaining, sensitivity and specificity were 67 % and 82 %; while positive and negative predictive values were 73 % and 78 %, respectively. When combined, there was improvement in sensitivity (75 %) with specificity of 82 %. The overall accuracy for the combined tests (80 %) was modestly higher than either IHC or molecular alone (76 % and 78 %)
4 Discussion
The distinction between benign and malignant parathyroid tumors can be difficult in the absence of obvious malignant characteristics, such as local recurrence or distant metastasis. Indeed, in a large series of metastatic CA cases, nearly half were initially classified as adenomas [31]. The high prevalence of CDC73 mutations in CA [10, 13, 14] led to suggestions that genetic screening may be an important clinical tool for improving diagnostic accuracy.
It has also been suggested that immunostaining of parafibromin should be considered a criterion for malignancy. The more common mutations of CDC73 gene are truncating and are most often accompanied by loss of parafibromin immunostaining. Indeed, several studies [11, 12, 14, 18, 21] indicate a high specificity for loss of parafibromin staining as a criterion in diagnosing carcinoma, since only one of some hundreds of unselected parathyroid adenomas was similarly IHC-negative. Consequently, loss of parafibromin staining has been advanced as a key index of malignant behaviour [12].
Our carcinoma series confirms a high prevalence (73 %) of CDC73 mutations in apparently sporadic tumors, with the expected range of clinical outcomes. Of the 15 cases, 11 were still alive after a median follow-up of 83 months (range 30–221 months). For the three whose cause of death was known, all had metastatic disease, all had mutations, and all were IHC-negative. In the additional single living patient with metastasis, she had a CDC73 mutation and her tumor was also IHC-negative.
In patients whose disease was locally confined, the genetic and IHC findings were more heterogeneous. Of the 11 subjects so classified, 6 had a mutation and 4 were IHC-negative (see Table 1). The number of IHC-positive carcinomas (4 of total 12) is in line with the recent observations of DeLellis [7]. The fact that all IHC patterns showing some significant degree of immunopositivity were classified as immunopositive, likely contributes to the overall rate, but the size of this effect is not known, and the assessment subjective. A consensus-driven IHC scoring system would be welcome.
Among the histologically typical adenomas, we identified CDC73 gene lesions in 18 %. It is not surprising that we did not identify constitutional mutations, but the same cannot be said of IHC negativity. It has been suggested that sporadic adenoma is never essentially “negative” at immunostaining, but we identified three cases that we considered unequivocally IHC-negative (Fig. 4c and d). In one case, a relative was affected by PHPT so we speculate that the CDC73 mutation was not identified, and further analysis is required. The other two with immunonegativity (#TA2 and #TA8) are more difficult to explain but the fact that they both showed deleterious somatic CDC73 mutations suggests that the tumor phenotype can vary significantly in response to mutation and that immunostaining will indeed be negative in a small but significant non-zero proportion of classical adenomas.
We also note that LOH at the CDC73 locus was observed in 10/46 cases (see Supplementary Material). The frequency was similar in the TA group (3/17) as in AA or CA subjects (3/14 and 4/15, respectively). In no instance did the LOH region span the entire range of markers (D1S215 to D1S413, ~ 20 megabases). Somatic point mutations, consistent with the Knudson 2-hit hypothesis, were found in 3 of the LOH positive cases, but our methods would only detect those somatic mutations that led to small sequence changes in CDC73 exons or intron-exon boundaries that were amplifiable by standard PCR techniques and Sanger sequencing. Furthermore, deletions that are too big to be detected by PCR but still too small to be detected by LOH using our panel of markers would also be missed, although such mutations are uncommon [32]. Another explanation for the absence of matching second hits is the possibility that large-scale hemizygous deletions by themselves may be tumorigenic [33]. Evidence is accumulating that deletion-derived haploinsufficiency at chromosomal loci with an excess of those genes down-regulating cell proliferation results in clonal selection of cells that proliferate faster, thereby creating a cell population predisposed to downstream tumorigenic events other than mutation or large-scale deletion, but resulting in homozygous inactivation of the locus. Better understanding of deletions in the CDC73 region (1q31.2) would be an important first step in the exploration of this hypothesis.
Similarly, LOH was not detected in 4 of 9 cases with somatic mutations. Two of those cases are readily explained by the presence of a germline point mutation, but the others may be attributable to the methodological limitations noted above. LOH, using the standard marker panels, did not appear to contribute to the diagnosis or management of our patients. However, more detailed studies using methods such as comparative genomic hybridization and exomic sequencing may provide important new clues to unanswered questions about the phenotype associated with CDC73 mutations [32].
In our series, parafibromin immunostaining showed a sensitivity of 67 % and a specificity of 82 % in diagnosing carcinoma, both somewhat lower than others have reported [16, 18]. Since typical adenomas are much more frequent than carcinomas, immunostaining can be regarded as only one tool for assessing malignancy.
One of the purposes of this study was to clarify the clinical, genetic and immunohistochemical characteristics distinguishing atypical adenoma from typical adenoma on one hand and parathyroid carcinoma on the other. While the hypothesis that atypical adenoma may be a precursor to carcinoma remains an attractive one, there is little direct evidence for this progression. As an intermediate form of parathyroid tumor in terms of typical metaplastic features (mitotic index, partial capsular invasion), the large group of atypical adenomas we studied here was also intermediate with respect to frequency of CDC73 mutation and IHC negativity. Clinical progression was surprisingly low. However, it was not much different from the experience of DeLellis [7], who found no recurrence in 24 AA subjects after a mean of 8 years follow-up. Our one exception was an individual belonging to a 4-generation family with multiple affected members carrying a germline truncating mutation. The details of that family have been reported previously [25].
Looking at all three types of parathyroid tumors in our cohort, we suggest that careful clinical follow-up, accurate histopathologic classification, reliable immunohistochemical staining for parafibromin and a comprehensive search for somatic and germline CDC73 mutations may all be important requisites for appropriate management of HRPT2 related tumors. In the case of typical adenomas, clinical assessment and routine histopathology are almost always sufficient for diagnosis and appropriate disposition. The key exception remains a positive family history, or the presence of other tumors suggestive of HPT-JT syndrome.
In contrast, genetic analysis (tumor and germline DNA) is probably warranted in most atypical adenoma cases, but the role of immunohistochemistry is less certain. Indeed the presence of somatic mutations could suggest the diagnosis of carcinoma and/or a progression of an AA in a carcinoma; however there are no data demonstrating that the presence of a CDC73 mutation is predictive for a more aggressive behavior.
Longer follow-up is needed to assess whether this tumor type progresses to frank neoplasia. In suspected parathyroid carcinoma, the usual clinical characteristics (tumor dimension, stony consistency and progressive hypercalcemia) are hardly sufficient for diagnosis and specific histopathologic characteristics (total capsular invasion and/or vessel invasion and/or distant metastasis and/or local recurrence) should be present to establish a diagnosis.
Since most parathyroid carcinomas are the result of parafibromin deficiency, genetic analysis of genomic DNA and search for somatic mutation of CDC73 gene are recommended. The importance of such a genetic analysis lies in the expectation that screening of asymptomatic carriers in the affected family will lead to effective early intervention [10, 25, 34]. This also applies to the proband in relation to the risk of recurrence, and to the increased risk to carriers of other CDC73-related tumors [26].
Finally the recommendation regarding the diagnostic properties of immunostaining alone needs to be reconsidered. Although initially stated to be highly specific, IHC-negativity may be observed in typical adenomas (as we show here), and in atypical adenoma [16]. It seems likely that some combination of specific immunohistochemistries [14, 19, 35] may improve sensitivity and specificity. In the interim, IHC may be considered most useful for special situations where classification is uncertain.
Our study confirms that truncating constitutional mutations of CDC73 are common in sporadic carcinoma. Moreover, we observed a recurrent mutation in exon 7, which may be a genetic hot spot, since it is seen in both germline and somatic DNA.
In conclusion our data suggest that parafibromin immunostaining cannot replace molecular analysis as a means of materially improving the accuracy of a diagnosis of parathyroid malignancy. In our cohort, CDC73 gene lesions or loss of nuclear immunostaining of parafibromin was not associated with a malignant behaviour of parathyroid AA followed up, on average, for 6 years. Combined molecular and immunohistochemistry testing appears to be the most effective approach to the differential diagnosis of parathyroid tumors.
References
S.J. Marx, Hyperparathyroid and hypoparathyroid disorders. N. Engl. J. Med. 343, 1863–1865 (2000)
J.M. Sharretts, W.F. Simonds, Clinical and molecular genetics of parathyroid neoplasms. Best Pract. Res. Clin. Endocrinol. Metab. 24, 491–502 (2010)
C. Marcocci, F. Cetani, M.R. Rubin, S.J. Silverberg, A. Pinchera, J.P. Bilezikian, Parathyroid carcinoma. J. Bone Miner. Res. 23, 1869–1880 (2008)
J.M. Sharretts, E. Kebebew, W.F. Simonds, Parathyroid cancer. Semin. Oncol. 37, 580–590 (2010)
B. Schantz, B. Castleman, Parathyroid carcinoma. A study of 70 cases. Cancer 31, 600–605 (1973)
L. Bondeson, L. Grimelius, R.A. DeLellis, R. Lloyd, G. Akerstrom, C. Larsson, A. Arnold, C. Eng, E. Shane, J.P. Bilezikian, Parathyroid carcinoma, in Pathology and genetics of tumours of endocrine organs. WHO classification of tumours, ed. by R.A. DeLellis, R.V. Lloyd, P.U. Heitz, C. Eng (IARC Press, Lyon, 2004), pp. 124–127
R.A. DeLellis, Challenging lesions in the differential diagnosis of endocrine tumors: parathyroid carcinoma. Endocr. Pathol. 19, 221–225 (2008)
J.D. Carpten, C.M. Robbins, A. Villablanca, L. Forsberg, S. Presciuttini, J. Bailey-Wilson, W.F. Simonds, E.M. Gillanders, A.M. Kennedy, J.D. Chen, S.K. Agarwal, R. Sood, M.P. Jones, T.Y. Moses, C. Haven, D. Petillo, P.D. Leotlela, B. Harding, D. Cameron, A.A. Pannett, A. Höög, H. Heath 3rd, L.A. James-Newton, B. Robinson, R.J. Zarbo, B.M. Cavaco, W. Wassif, N.D. Perrier, I.B. Rosen, U. Kristoffersson, P.D. Turnpenny, L.O. Farnebo, G.M. Besser, C.E. Jackson, H. Morreau, J.M. Trent, R.V. Thakker, S.J. Marx, B.T. Teh, C. Larsson, M.R. Hobbs, HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat. Genet. 32, 676–680 (2002)
P.J. Newey, M.R. Bowl, T. Cranston, R.V. Thakker, Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum. Mutat. 31, 295–307 (2010)
T.M. Shattuck, S. Valimäki, T. Obara, R.D. Gaz, O.H. Clark, D. Shoback, M.E. Wierman, K. Tojo, C.M. Robbins, J.D. Carpten, L.O. Farnebo, C. Larsson, A. Arnold, Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N. Engl. J. Med. 349, 1722–1729 (2003)
C.C. Juhlin, A. Villablanca, K. Sandelin, F. Haglund, J. Nordenstrom, L. Forsberg, L. Branstrom, T. Obara, A. Arnold, C. Larsson, A. Höög, Parafibromin immunoreactivity: its use as an additional diagnostic marker for parathyroid tumor classification. Endocr. Relat. Cancer 14, 501–512 (2007)
F. Cetani, E. Ambrogini, P. Viacava, E. Pardi, G. Fanelli, A.G. Naccarato, S. Borsari, M. Lemmi, P. Berti, P. Miccoli, A. Pinchera, C. Marcocci, Should parafibromin staining replace HRPT2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? Eur. J. Endocrinol. 156, 547–554 (2007)
V.M. Howell, C.J. Haven, K. Kahnoski, S.K. Khoo, D. Petillo, J. Chen, G.J. Fleuren, B.G. Robinson, L.W. Delbridge, J. Philips, A.E. Nelson, U. Krause, K. Hammje, H. Dralle, C. Hoang-Vu, O. Gimm, D.J. Marsh, H. Morreau, B.T. Teh, HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J. Med. Genet. 40, 657–663 (2003)
V.M. Howell, A. Gill, A. Clarkson, A.E. Nelson, R. Dunne, L.W. Delbridge, B.G. Robinson, B.T. Teh, O. Gimm, D.J. Marsh, Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. J. Clin. Endocrinol. Metab. 94, 434–441 (2009)
L.J. Krebs, T.M. Shattuck, A. Arnold, HRPT2 mutational analysis of typical sporadic parathyroid adenomas. J. Clin. Endocrinol. Metab. 90, 5505–5507 (2005)
F. Cetani, E. Pardi, S. Borsari, P. Viacava, G. Dipollina, L. Cianferotti, E. Ambrogini, E. Gazzerro, G. Colussi, P. Berti, P. Miccoli, A. Pinchera, C. Marcocci, Genetic analysis of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J. Clin. Endocrinol. Metab. 89, 5583–5591 (2004)
M.H. Tan, C. Morrison, P. Wang, X. Yang, C.J. Haven, C. Zhang, P. Zhao, M.S. Tretiakova, E. Korpi-Hyovalti, J.R. Burgess, K.C. Soo, W.K. Cheah, B. Cao, J. Resau, H. Morreau, B.T. Teh, Loss of parafibromin immunoreactivity is a distinguishing feature of parathyroid carcinoma. Clin. Cancer Res. 10, 6629–6637 (2004)
A.J. Gill, A. Clarkson, O. Gimm, J. Keil, H. Dralle, V.M. Howell, D.J. Marsh, Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidism-jaw tumor (HPT-JT) syndrome-related adenomas from sporadic parathyroid adenomas and hyperplasias. Am. J. Surg. Pathol. 30, 1140–1149 (2006)
C.C. Juhlin, I.-L. Nilsson, K. Johansson, F. Haglund, A. Villablanca, A. Höög, C. Larsson, Parafibromin and APC as screening markers for malignant potential in atypical parathyroid adenomas. Endocr. Pathol. 21, 166–177 (2010)
S. Mangray, K.C. Kurek, E. Sabo, R.A. DeLellis, Immunohistochemical expression of parafibromin is of limited value in distinguishing parathyroid carcinoma from adenoma. Mod. Pathol. 21, 108A (2008). abstract
G.G. Fernandez-Ranvier, E. Khanafshar, D. Tacha, M. Wong, E. Kebebew, Q.Y. Duh, O.H. Clark, Defining a molecular phenotype for benign and malignant parathyroid tumors. Cancer 115, 334–344 (2009)
C.C. Juhlin, C. Larsson, T. Yakoleva, I. Leibiger, B. Leibiger, A. Alimov, G. Weber, A. Hoog, A. Villablanca, Loss of parafibromin expression in a subset of parathyroid adenomas. Endocr. Relat. Cancer 13, 509–523 (2006)
C.J. Haven, M. van Puijenbroek, M.H. Tan, B.T. Teh, G.J. Fleuren, T. van Wezel, H. Morreau, Identification of MEN1 and HRPT2 somatic mutations in paraffin-embedded (sporadic) parathyroid carcinomas. Clin. Endocrinol. 67, 370–376 (2007)
V. Guarnieri, L. Canaff, F.H.J. Yun, A. Scillitani, C. Battista, L.A. Muscarella, B.Y.L. Wong, A. Notarangelo, L. D’Agruma, M. Sacco, D.E.C. Cole, G.N. Hendy, Calcium-sensing receptor (CASR) mutations in hypercalcemic states: studies from a single endocrine clinic over three years. J. Clin. Endocrinol. Metab. 95, 1819–1829 (2010)
V. Guarnieri, A. Scillitani, L.A. Muscarella, C. Battista, N. Bonfitto, M. Bisceglia, S. Minisola, M.L. Mascia, L. D’Agruma, D.E.C. Cole, Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J. Clin. Endocrinol. Metab. 91, 2827–2832 (2006)
V. Guarnieri, M. Bisceglia, N. Bonfitto, F. Cetani, C. Marcocci, S. Minisola, C. Battista, I. Chiodini, D.E.C. Cole, A. Scillitani, Re: Familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery 144, 839–840 (2008)
L. Grimelius, R.A. DeLellis, L. Bondeson, G. Akerstrom, A. Arnold, K.O. Frinssila, G.N. Hendy, D. Dupuy, C. Eng, Parathyroid adenoma, in Pathology and genetics of tumours of endocrine organs. WHO classification of tumours, ed. by R.A. DeLellis, R.V. Lloyd, P.U. Heitz, C. Eng (IARC Press, Lyon, 2004), pp. 128–132
P. Parrella, D. Sidransky, S.L. Merbs, Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 59, 3032–3037 (1999)
G. Masi, L. Barzon, M. Iacobone, G. Viel, A. Porzionato, V. Macchi, R. De Caro, G. Favia, G. Palù, Clinical, genetic, and histopathologic investigation of CDC73-related familial hyperparathyroidism. Endocr. Relat. Cancer 15, 1115–1126 (2008)
R.I. Skotheim, C.B. Diep, S.M. Kraggerud, K.S. Jakobsen, R.A. Lothe, Evaluation of loss of heterozygosity/allelic imbalance scoring in tumor DNA. Cancer Genet. Cytogenet. 127, 64–70 (2001)
K. Sandelin, O. Tullgren, L.O. Farnebo, Clinical course of metastatic parathyroid cancer. World J. Surg. 18, 594–599 (1994)
R. Domingues, R.A. Tomaz, C. Martins, C. Nunes, M.J. Bugalho, B.M. Cavaco, Identification of the first germline HRPT2 whole-gene deletion in a patient with primary hyperparathyroidism. Clin. Endocrinol. 76, 33–38 (2012)
C.D. Greenman, Haploinsufficient gene selection in cancer. Science 337, 47–48 (2012)
T.G. Kelly, T.M. Shattuck, M. Reyes-Mugica, A.F. Stewart, W.F. Simonds, R. Udelsman, A. Arnold, T.O. Carpenter, Surveillance for early detection of aggressive parathyroid disease: carcinoma and atypical adenoma in familial isolated hyperparathyroidism associated with a germline HRPT2 mutation. J. Bone Miner. Res. 21, 1666–1671 (2006)
C.C. Juhlin, F. Haglund, T. Obara, A. Arnold, C. Larsson, A. Höög, Absence of nucleolar parafibromin immunoreactivity in subsets of parathyroid malignant tumours. Virchows Arch. 459, 47–53 (2011)
V.M. Howell, J.W. Cardinal, A.L. Richardson, O. Gimm, B.G. Robinson, D.J. Marsh, Rapid mutation screening for HRPT2 and MEN1 mutations associated with familial and sporadic primary hyperparathyroidism. J. Mol. Diagn. 8, 559–566 (2006)
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by grants from Ministero della Salute of Italy, Ricerca Oncologica “RF-OGR-2006_416850” (to A.Sc), from Ministero della Salute of Italy, Progetti di Ricerca di Interesse Nazionale—cofinanziamento 2007 (PRIN 2007) (to S.M.), from Istituto Superiore di Sanità “ITALIA-USA Program 2007—Malattie Rare” grant no. 8900000 (to A.Sp.) and grants from Ministero della Salute of Italy (Ricerca Corrente 2003 to A.Sc. and 2007 to M.C.) and a fellowship by FIRC 2006 (VG).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 151 kb)
Rights and permissions
About this article
Cite this article
Guarnieri, V., Battista, C., Muscarella, L.A. et al. CDC73 mutations and parafibromin immunohistochemistry in parathyroid tumors: clinical correlations in a single-centre patient cohort. Cell Oncol. 35, 411–422 (2012). https://doi.org/10.1007/s13402-012-0100-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-012-0100-x