
Abstract
Parathyroid neoplasms encompass a spectrum of proliferative lesions that include adenomas, atypical adenomas, and carcinomas. While the diagnosis of adenomas is usually straightforward, parathyroid carcinomas (PTCAs) often present considerable diagnostic challenges. Fibrosis and mitotic activity are common in PTCAs, but these features are not specific for malignancy. An unequivocal diagnosis of PTCA should be restricted to those tumors that invade adjacent soft tissues, thyroid gland, blood vessels, or perineural spaces or to those cases with documented metastases. Atypical adenomas include those tumors that share some of the features of PTCA but lack evidence of invasive growth. A variety of genetic abnormalities, including HRPT2 mutations, occur in PTCAs. Mutations of the HRPT2 gene, which encodes parafibromin, are responsible for the development of the hyperparathyroidism–jaw tumor syndrome and have also been implicated in the development of a high proportion of sporadic PTCAs. Correlative immunohistochemical studies have revealed nuclear parafibromin immunoreactivity in adenomas but absence or partial loss of staining in PTCAs. While parafibromin immunohistochemistry represents an important step in the ability to diagnose PTCA, additional studies will be required to test the validity of this approach and to determine the roles of other genes in the development of these tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
General Considerations
Primary hyperparathyroidism (P-HPT) is now recognized as one of the most common of all endocrine disorders [1]. When defined by clinical and pathological parameters, solitary adenomas account for 80–85% of cases of P-HPT, hyperplasias for 10–15%, and carcinomas for less than 1% in the USA and most Western European countries [2, 3]. However, the prevalence of parathyroid carcinoma has been reported to be as high as 5% in Italy and Japan. Whether the relatively high prevalence of parathyroid carcinoma in these countries is the result of true geographic differences or is related to differing pathological criteria is unknown.
The literature in surgical pathology is replete with examples of difficult diagnostic issues involving the distinction of benign and malignant endocrine tumors and the parathyroid glands are no exception to these diagnostic dilemmas. In particular, the distinction of parathyroid carcinomas from other proliferative lesions of these glands including some forms of hyperplasia and adenomas, is challenging. As noted by Edgar Norris (1948), “In 1926, Herxheimer and in 1933 Jaffe debated the existence of primary carcinoma of the parathyroids, and even today, in the minds of many, the question as to whether malignant epithelial tumors arise in the parathyroids is debatable” [4]. In that same paper, Norris reviewed 44 cases of parathyroid carcinoma reported between 1907 and 1945 and concluded that only 15 of the cases satisfied his criteria for malignancy.
Norris stressed the presence of a thick fibrous capsules with intratumoral fibrous septa, invasion of adjacent tissues, and vascular invasion as criteria for malignancy [4]. He further noted that “there is no definitely dependable single characteristic by which a parathyroid carcinoma may be diagnosed clinically or pathologically. The diagnosis can be made only by utilizing all of the information and by taking account of the known variations in the biologic behavior and structural constitution of this lesion”.
In the first edition of the AFIP Fascicle of Tumors of the Parathyroid Glands, Castleman noted that “if enough sections are taken through the capsule, in most cases evidence of capsular invasion will be evident. This in itself, however, is inadequate evidence for a diagnosis of cancer…there is a great tendency in these tumors to trabecular arrangement, which is often exaggerated by the cylindrical shape of the cells. Mitoses, which are absent in the adenoma, are always present in the carcinomas” [5].
Schantz and Castleman in a classic paper on the pathology of parathyroid carcinoma established the major criteria for the diagnosis of this tumor type [6]. They reported mitotic activity in 80% of carcinomas; however, later publications demonstrated that mitotic activity was also present in at least 60% to 70% of parathyroid adenomas [7]. Bondeson and coworkers noted that “the presence of scattered foci of coagulative necrosis plus macronucleoli and mitotic activity in excess of 5 mitoses per 50 high power fields constitutes a triad indicating a high risk of malignant behavior” [2]. It should be emphasized, however, that a significant proportion of parathyroid tumors without this extent of mitotic activity may pursue a malignant course. Atypical mitoses, on the other hand, appear to be a specific feature of carcinomas. Capsular invasion was present in at least 60% of Schantz and Castleman’s series of carcinomas and was characterized by extension of tumor through the tumor capsule into the surrounding adipose tissue, skeletal muscle, or thyroid gland [6]. True capsular invasion should be distinguished from entrapment of tumor cells within the capsule, which may be particularly prominent in adenomas that have undergone cystic degenerative changes.
Vascular invasion was present in only 10–15% of carcinomas in Schantz and Castleman’s series. The diagnosis of vascular invasion should be restricted to those cases in which the affected vessels are present within the tumor capsule or within vessels in surrounding soft tissues. The tumor clusters should be surrounded by endothelium or associated with fibrin thrombi. Artifactual dislodgement of tumor cells is characterized by the presence of irregularly shaped cell clusters that are not endothelialized or associated with thrombus. Although vascular invasion is an uncommon feature of parathyroid carcinomas, it is virtually diagnostic of malignancy. Perineural space invasion is also uncommon but, similar to vascular invasion, is diagnostic of malignancy.
Parathyroid carcinomas are usually larger than adenomas and are often adherent to the adjacent thyroid gland or soft tissues of the neck [2]. The latter feature should alert the surgeon to the possibility of malignancy and should lead to consideration of an en bloc resection. However, it is important to recognize that very large adenomas are frequently cystic with prominent areas of peritumoral fibrosis and varying degrees of adherence to the surrounding tissues. The value of frozen section in these situations is questionable since the distinction of a carcinoma from a degenerated adenoma may be impossible.
Most carcinomas have a solid growth pattern with tumor cells arranged in diffuse masses, small nests, or trabeculae. A few tumors may exhibit spindle cell, follicular, and even papillary patterns. Many carcinomas exhibit mild variation in nuclear size and shape and may be indistinguishable from adenomas; however, some tumors may exhibit profound pleomorphism with coarse chromatin and macronucleoli, as noted by Bondeson et al. [2]. This feature should be distinguished from the nuclear atypia encountered so frequently in parathyroid adenomas and other benign endocrine tumors (so-called endocrine atypia) [8].
Implantation of parathyroid tissue may occur following incomplete excision of hyperplastic parathyroid tissue, a condition that has been termed “parathyromatosis” [9]. A similar phenomenon may occur as a result of intraoperative capsular rupture or incomplete excision of a parathyroid adenoma. Differentiation of parathyromatosis from recurrence of a previously resected and possibly underdiagnosed carcinoma is exceedingly difficult since both may be associated with extensive infiltration of soft tissues and fibrosis. The presence of vascular and perineural space invasion are helpful features to distinguish recurrent parathyroid carcinoma from parathyromatosis.
Atypical Adenomas
The term “atypical adenoma” has been used to describe a subset of parathyroid tumors that share some of the features of carcinomas (fibrosis, mitoses, questionable capsular invasion) but which lack definitive evidence of invasive growth [10]. In this regard, the tumors are similar to those described by Bondeson et al. as “equivocal” [11]. In a study of 24 tumors classified as atypical adenomas by Guiter and DeLellis, the mean tumor size was 2.2 cm and mean tumor weight was 6.5 g [12]. The most common findings in this group were entrapment of tumor within the surrounding capsules (87%), intratumoral fibrosis (75%), prominent hemosiderin deposits (58%), cyst formation (50%), mitoses (30%), and peritumoral fibrosis (25%). Capsular invasion without extension of tumor beyond the capsule and intratumoral vascular invasion were each present in single cases but none of the cases had evidence of necrosis. The average clinical follow-up is now 8 years and none of the patients has developed tumor recurrence or metastatic disease. The findings in this study suggest that the behavior of atypical adenomas, as defined in this study, does not differ from that of adenomas of usual type [12]. However, close clinical follow-up is recommended because of the prolonged natural history of many parathyroid carcinomas.
Studies of the proliferative fractions of parathyroid tumors have revealed higher values in carcinomas than in adenomas, but the overlap of values in equivocal cases has limited the value of this approach [13]. An additional approach has involved the use of antibodies to p27, which encodes a cyclin-dependent kinase inhibitor [14]. As compared with adenomas, carcinomas had a 3-fold decrease in p27 expression. These findings have suggested that p27 and Ki-67 labeling indices may be helpful in the distinction of parathyroid adenomas (↓ Ki-67, ↑p27) and carcinomas (↑ Ki-67, ↓p27). Stojadinovic and coworkers have reported that the phenotype p27 (+), bcl-2 (+), Ki-67 (−), and mdm2 (+) was present in 76% and 29% of typical adenomas and atypical adenomas, respectively, and in no cases of parathyroid carcinoma [15].
Molecular Approaches
Comparative genomic hybridization (CGH) analyses have demonstrated losses of 1p and 13q in more than 40% of cases of parathyroid carcinoma [16]. Common regions of loss include 1p21–22 (41%), 13q14–q31 (41%), 9p21-pter (28%), 6q22–q24 (24%), and 4q24 (21%) while regions of gain include 19p (45%), Xcq13 (28%), 9q33qter (24%), 1q31–q32 (21%), and 16p (21%). Kytola et al. suggested a model based on these observations characterized by early gains of Xq and 1q followed by loss of 13q, 9p, and 1p and gain of 19p [16]. Losses of 1p, 4q, and 13q and gains of 1q, 9q, 16p, 19p, and Xq were significantly more common in carcinomas than adenomas. Interestingly, loss of 11q13, the most common abnormality of adenomas, was undetectable in this study. This finding has suggested that adenomas developing along the MEN1 pathway have minimal potential for progression into carcinomas.
With fluorescence in situ by hybridization, Erickson et al. confirmed the lack of chromosome loss in carcinomas but noted chromosome gains in three of four patients who died of metastatic parathyroid carcinomas [17]. Haven et al., on the other hand, demonstrated 1q and 11q LOH in 55% and 50% of parathyroid carcinomas with combined losses in 36% [18]. Valimaki and colleagues demonstrated 1p LOH in 43% of adenomas; 60% of these cases harbored alterations of either 1p, 11q13, or both [19]. Loss of heterozygosity in sporadic adenomas occurred frequently on the distal part of 1q whereas deletions in 1p in carcinomas occurred more proximally. These observations suggest that 1p may harbor at least two different suppressor genes that may be involved in parathyroid tumorigeneses.
Several groups have demonstrated LOH of 13q, a region that includes RB and BRCA2, in parathyroid carcinomas. In the series reported by Cryns et al., 11of 11 specimens from patients with parathyroid carcinoma and one of 19 adenomas lacked an RB allele [20]. Correlative immunohistochemical studies demonstrated a complete or predominant absence of RB expression in carcinomas whereas none of the adenomas had abnormal RB staining patterns. BRCA2 has also been suggested as a potential suppressor gene in these tumors [21]. However, the contribution of both RB and BRCA2 to the development of carcinomas has been controversial. In a recent study by Cetani et al., LOH for at least one marker of the RBI locus was found in six of six carcinomas whereas LOH for BRCA2 was found in three of five cases [22]. In the same series, LOH for RB and BRCA2 was demonstrated in 28.8% and 17.4%, respectively, of adenomas.
Shattuck et al. recently performed direct sequencing of RB and BRCA2 in parathyroid carcinomas that showed LOH at the RB locus and/or 13q loss by CGH and were unable to find microdeletions, insertions, or point mutations of either gene [23]. They concluded that neither RB nor BRCA2 were likely to act as tumor suppressor genes in carcinomas. However, these results do not exclude the possibility that the decreased RB function in carcinomas, whether secondary or because of epigenetic effects, may play a role in tumor development. It is also possible that other genes on chromosome 13 may be implicated in the development of parathyroid carcinomas.
Studies of heritable tumor syndromes have provided considerable insight into the molecular basis of the corresponding sporadic tumors. Mutations of the HRPT2 gene are responsible for the development of the hyperparathyroidism–jaw tumor (HPT–JT) syndrome, which is inherited as an autosomal dominant trait [24]. The commonest manifestations of this syndrome include primary hyperparathyroidism, fibro-osseous lesions of the mandible and maxilla, and a variety of renal lesions [25]. Hyperparathyroidism occurs as a result of neoplasms of one or more parathyroid glands, which frequently show cystic change. Interestingly, parathyroid carcinomas occur in 10–15% of patients with this syndrome.
The role of the HRPT2 gene in the pathogenesis of sporadic parathyroid carcinomas was first demonstrated by Howell et al. in 2003 [26]. Subsequent studies by Shattuck and coworkers demonstrated that parathyroid carcinomas from ten of 15 patients had HRPT2 mutations which were predicted to inactivate the encoded parafibromin protein [27]. Importantly, the HRPT2 mutations in three of the parathyroid carcinomas of these patients were identified as germline mutations. The latter finding suggests that a subset of patients with apparent sporadic parathyroid carcinomas carry germline mutations in the HRPT2 gene and may, in fact, have the HPT–JT syndrome or a variant of that syndrome. On the basis of these findings, it has been suggested that all patients with parathyroid carcinoma should have jaw and renal imaging studies and also should be tested for germline HRPT2 mutations.
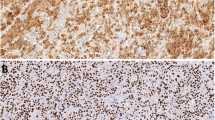
Loss of parafibromin was first reported as a molecular marker for parathyroid carcinoma by Tan and colleagues [28]. These workers noted that loss of parafibromin nuclear staining had a 96% sensitivity and 99% specificity for the definitive diagnosis of parathyroid carcinoma. In addition to parafibromin loss in carcinomas, this protein was also absent from HPT–JT associated adenomas. Similar results have been reported by other groups, although the different studies employed different scoring systems [29–31].
Juhlin et al. have demonstrated that 68% of unequivocal carcinomas exhibited reduced expression of parafibromin while 100% of adenomas were positive [32]. Moreover, three of six carcinomas with known HRPT2 mutations had reduced expression of parafibromin. They conclude that parafibromin immunohistochemistry could be used as an additional marker for parathyroid tumor classification with parafibromin-positive cases having a low risk of malignancy and cases with reduced protein expression representing either carcinomas or adenomas with HRPT2 mutations [32]. Of particular interest is the observation that four of five metastatic parathyroid carcinomas observed in patients with chronic renal failure were positive for parafibromin. This finding suggests that genetic events other than HRPT2 mutations may be of significance in the genesis of different subsets of PTCA [33].
In our own experience [34, 35], loss of parafibromin staining has been noted in a subset of adenomas unassociated with the HPT–JT syndrome while some carcinomas have had positive staining. These observations emphasize the need for the use of well-characterized parafibromin antibodies as well as standardized fixation and retrieval conditions, staining protocols, and scoring systems before this approach becomes the standard of practice. While parafibromin immunohistochemistry represents an important step in the ability to diagnose PTCA, additional studies will be required to test the validity of this approach and to determine the roles of other genes in the development of these tumors.
References
DeLellis RA, Mazzalia P, Mangray S. Primary hyperparathyroidism: a current perspective. Arch Pathol Lab Med 132:1251–62, 2008.
Bondeson L, Grimeluis L, DeLellis RA, et al. Parathyroid carcinoma. In: DeLellis RA, Lloyd RV, Heitz PN, Eng C, eds. Pathology and genetics of tumours of endocrine organs (WHO classification). Lyon: IARC, pp. 224–7, 2004.
DeLellis RA. Parathyroid carcinoma. An overview. Adv Anat Pathol 12:53–1, 2005. doi:10.1097/01.pap.0000151319.42376.d4.
Norris EH. Collective review. Carcinoma of the parathyroid glands with a preliminary report of 3 cases. Int Abstracts of Surg 86:1–21, 1948.
Castleman B. Tumors of the Parathyroid Glands. Atlas of Tumor Pathology. Section IV—Fascicle 15. Armed Forces Institute of Pathology. Washington, DC 1952.
Schantz A, Castleman B. Parathyroid carcinoma. A study of 70 cases. Cancer 31:600–5, 1973. doi:10.1002/1097-0142(197303)31:3<600::AID-CNCR2820310316>3.0.CO;2-0.
Snover D, Foucar K. Mitotic activity in benign thyroid diseases. Am J Clin Pathol 75:345–7, 1981.
DeLellis RA. Tumors of the parathyroid glands. Atlas of tumor pathology, 3rd series, fascicle 6. Washington DC: Armed Forces Institute of Pathology, 1993.
Reddick RL, Costa J, Marx SJ. Parathyroid hyperplasia and parathyromatosis. Lancet 1:549, 1977. doi:10.1016/S0140-6736(77)91414-3.
Levin KE, Chew KL, Ljung BM. Deoxyribonucleic acid cytometry helps identify parathyroid carcinomas. J Clin Endocrinol Metab 67:770–4, 1988.
Bondeson L, Sandelin K, Grimeluis L. Histopathological variables and DNA cytometry in parathyroid carcinoma. Am J Surg Pathol 17:820–9, 1993. doi:10.1097/00000478-199308000-00007.
Guiter GE, DeLellis RA. Risk of recurrence or metastasis in atypical parathyroid adenoma. Mod Pathol 15:115A, 2002, (Abstract).
Abbona GC, Papotti M, Gasparri G, Bussolati G. Proliferative activity in parathyroid tumors as detected by KI-67 immunostaining. Hum Pathol 26:135–8, 1995. doi:10.1016/0046-8177(95)90028-4.
Erickson LA, Jin L, Wollan P, Thompson GB, van Heerden LA, Lloyd RV. Parathyroid hyperplasia, adenomas and carcinoma: differential expression of p27kip1 protein. Am J Surg Pathol 23:288–5, 1999. doi:10.1097/00000478-199903000-00007.
Stojadinovic A, Hoos A, Nissan A, et al. Parathyroid neoplasms; clinical, histopathological and tissue microarray-based molecular analysis. Hum Pathol 34:54–4, 2003. doi:10.1053/hupa.2003.55.
Kytola S, Farnebo F, Obara T, et al. Patterns of chromosomal imbalances in parathyroid carcinomas. Am J Pathol 157:570–6, 2000.
Erickson LA, Jalal SM, Harwood A, Sherer B, Jin L, Lloyd RV. Analysis of parathyroid neoplasms by interphase fluorescence in situ hybridization. Am J Surg Pathol 38:578–4, 2004. doi:10.1097/00000478-200405000-00003.
Haven CJ, van Puijenbroek M, Karperien M, Fleuren GJ, Morreau H. Differential expression of the calcium sensing receptor and combined loss of chromosome 1p and 11q in parathyroid carcinoma. J Pathol 202:86–4, 2004. doi:10.1002/path.1489.
Valimaki S, Forsberg L, Farnebo LO, Larsson C. Distinct target regions for chromosome 1p deletions in parathyroid adenomas and carcinomas. Int J Oncol 21:727–5, 2002.
Cryns VT, Thor A, Xu HJ. Loss of the retinoblastoma tumor suppressor gene in parathyroid carcinoma. N Engl J Med 330:757–1, 1994. doi:10.1056/NEJM199403173301105.
Pearce SH, Trump D, Wooding C, Sheppard MN, Clayton RN, Thakker RV. Loss of heterozygosity studies at the retinoblastoma and breast cancer susceptibility BRCA2 loci in pituitary, parathyroid, pancreatic and carcinoid tumors. Clin Endocrinol (Oxf) 45:195–200, 1996. doi:10.1046/j.1365-2265.1996.d01-1561.x.
Cetani F, Pardi E, Viacava P, et al. A reappraisal of the Rb1 gene abnormalities in the diagnosis of parathyroid carcinoma. Clin Endocrinol (Oxf) 60:99–106, 2004. doi:10.1111/j.1365-2265.2004.01954.x.
Shattuck TM, Kim TS, Costa J, et al. Mutational analysis of RB and BRCA2 as candidate tumor suppressor genes in parathyroid carcinomas. Clin Endocrinol (Oxf) 59:180–9, 2003. doi:10.1046/j.1365-2265.2003.01814.x.
Carpten JD, Robbins CM, Villablanca A, et al. HRPT2 encoding parafibromin is mutated in hyperparathyroidism–jaw tumor syndrome. Nat Genet 32:676–80, 2002. doi:10.1038/ng1048.
Teh BT, Sweet KM, Morrison CD. Hyperparathyroidism–jaw tumor syndromeIn: DeLellis RA, Lloyd RV, Heitz PN, Eng C, eds. Pathology and genetics of tumours of endocrine organs (WHO classification).. Lyon: IARC, pp. 228–9, 2004.
Howell VM, Haven CJ, Kahnoski K, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumors. J Med Genet 40:657–3, 2003. doi:10.1136/jmg.40.9.657.
Shattuck TM, Valimaki S, Obara T, et al. somatic and germline mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med 349:1722–29, 2003. doi:10.1056/NEJMoa031237.
Tan MH, Morrison C, Wang P, et al. Loss of parafibromin immunoreactivity is a distinguishing feature of parathyroid carcinoma. Clin Cancer Res 10:6629–7, 2004. doi:10.1158/1078-0432.CCR-04-0493.
Juhlin C, Larsson C, Yakoleva T, et al. Loss of parafibromin expression in a subset of parathyroid adenomas. Endocr Relat Cancer 13:509–3, 2006. doi:10.1677/erc.1.01058.
Gill AJ, Clarkson A, Gimm O, et al. Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidism–jaw tumor associated adenomas from sporadic parathyroid adenomas and hyperplasias. Am J Surg Pathol 30:1140–49, 2006. doi:10.1097/01.pas.0000209827.39477.4f.
Cetani F, Ambrogini E, Viacava P, et al. Should parafibromin staining replace HRPT2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma. Europ J Endocrinol 156:547–4, 2007. doi:10.1530/EJE-06-0720.
Juhlin CC, Villablanca A, Sandelin K, et al. Parafibromin immunoreactivity; its use as an additional diagnostic marker for parathyroid tumor classification. Endocr Relat Cancer 14:501–2, 2007. doi:10.1677/ERC-07-0021.
Tominaga Y, Tsuzuki T, Matsuoka A, et al. Expression of parafibromin in distant metastatic parathyroid tumors in patients with advanced secondary hyperparathyroidism due to chronic kidney disease. World J Surg 32:815–1, 2008. doi:10.1007/s00268-007-9458-8.
Mangray S, Kurek KC, Sabo E, DeLellis RA. Immunohistochemical expression of parafibromin is of limited value in distinguishing parathyroid carcinoma from adenoma. Mod Pathol 21:108A, 2008, (abstract).
Mangray S, DeLellis RA. Parafibromin s a tool for the diagnosis of parathyroid tumors (letter). Adv Anat Pathol 15:179, 2008. doi:10.1097/PAP.0b013e3181709f83.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper was presented in part at the Annual Meeting of the Endocrine Pathology Society in March, 2008.
Rights and permissions
About this article
Cite this article
DeLellis, R.A. Challenging Lesions in the Differential Diagnosis of Endocrine Tumors: Parathryoid Carcinoma. Endocr Pathol 19, 221–225 (2008). https://doi.org/10.1007/s12022-008-9050-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-008-9050-2