
Abstract
Opiate abuse is a major global health problem, due in part to the fact that the HIV infection often occurs with intravenous drug abuse. There is strong clinical and preclinical evidence that opiate abuse promotes the neurodegeneration that can occur in association with HIV infection. Morphine or heroin can exert direct neurotoxic effects on neuronal cells and alter neuronal function. In addition, opiate administration after the virus infection has been established can exacerbate the neurotoxic properties of some of the HIV products. This can include the induction of pro-inflammatory mediators including both cytokines and chemokines and a loss of blood–brain barrier integrity. It is also clear that the activation of opioid receptors by agonists like morphine can initiate cross-talk interactions with other receptors, most notably the chemokine receptors CCR5 and CXCR4. Opiates clearly exert both pro- and anti-inflammatory activity, and our understanding of how these opposing influences are balanced in both the brain and periphery is rapidly advancing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
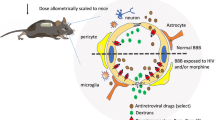


Keywords
- Opiate
- Morphine
- Heroin
- Neurodegeneration
- Blood–brain barrier
- HIV-1
- Co-receptor
- Heterologous desensitization
- Glutamate
- Chemokine
1 Introduction
Opiate drug abuse is a major contributing factor to the global AIDS epidemic. It is likely that over a third of the HIV infections in the USA can be linked to intravenous drug abuse, and global estimates suggest that almost 20 % of intravenous drug abusers are infected with HIV [1–4]. Chronic opioid abuse is a growing problem, due in part to the increase in the misuse of prescription opioid drugs in the USA [3]. Opioid abuse is associated with a decline in resistance to a number of opportunistic infections, and both direct and indirect processes are responsible for these immunosuppressive effects (reviewed in [5–7]). Work reported by a number of investigators, based on both clinical and laboratory research, has documented the capacity of heroin (or morphine) to inhibit adaptive and innate immune responses [5, 6, 8–10]. Moreover, experimental animal research shows that opioid administration leads to an increase in susceptibility to a number of infectious agents, including Candida albicans, Klebsiella pneumoniae, Streptococcus pneumoniae, herpes virus, murine leukemia virus, and Toxoplasma gondii [11–19]. However, the effect of chronic opioid administration on resistance to HIV infection is less clear. Experimental animal studies carried out with SIV have provided conflicting results, and this is almost certainly due, in part, to inconsistencies in the properties of the viral strain and the dose of morphine administered [20–23].
It is important to recognize that the immunosuppressive activity of the opioids is not universal, since it is clear that these drugs can have strong pro-inflammatory properties in certain circumstances. For example, current evidence suggests that opiates promote the neuropathology which can be associated with HIV infection by increasing the toxicity of some of the HIV proteins, particularly in the brain [24]. The neurotoxicity of the opiates is likely due to an elevation in the expression of pro-inflammatory cytokines in the brain [24, 25]. There is also evidence that opioid administration results in the degradation of the integrity of the blood–brain barrier (BBB), which may promote the exposure of the brain to additional pro-inflammatory cytokines which may more easily pass across the BBB [24, 26]. These studies are consistent with clinical evidence which suggests that opiate abuse leads to higher rates of encephalopathy in HIV-infected patients, compared to infected nondrug abusers [27, 28].
2 Opiates and Opioid Receptors
Opium, derived from the seedpods of Papaver somniferum, has been utilized for medicinal purposes since prehistoric times. Heroin, or diacetylmorphine, is chemically synthesized from opium and is one of the common opiates employed by intravenous drug abusers. Heroin is metabolized to morphine, 6-monoacetylmorphine, morphine-6-β-d-glucuronide, and morphine-3-β-d-glucuronide. While morphine is utilized widely as an analgesic and is the major bioactive heroin metabolite, several of the metabolites also possess opiate activity [29]. Endogenous opioid peptides, including endomorphins 1 and 2, leu- and met-enkephalin, and dynorphin, are produced in both the brain and in the periphery, and levels of opioid peptides appear to increase in response to inflammatory stimuli [30]. The regulation of endogenous peptide expression in the periphery by leukocytes is not well defined, and this information is important for our understanding of the regulation of the immune system by opioids.
There are three opioid receptors, and these are designated μ-, κ-, and δ-opioid receptors (MOR, KOR, and DOR) [5, 31]. Each of these receptors is expressed within the central and peripheral nervous system, although the relative expression may vary depending on the specific tissue site. The opioid receptors are also expressed by leukocytes, and molecular analysis of the opioid receptors expressed by leukocytes shows they are identical to those expressed in the CNS [32–34]. Experimental work on the modulation of the immune response is often conducted with one or more synthetic opioids, and these can have the advantage of being more receptor selective and/or possess a higher affinity for the respective opioid receptor type. The experimental use of these agonists can offer a major advantage in conducting experiments to understand the role of specific opioid receptors in the immune response, since opioid drugs of abuse are not highly receptor selective. For example, morphine has predominant binding activity for MOR but also activates both KOR and DOR. This means that the effects induced by morphine may be mediated by combinations of opioid receptor types. This can represent a critical issue since MOR and KOR can mediate opposing activities for cells of the immune system [35, 36].
3 Direct Mechanisms of Neurotoxicity of Opioids
The μ-opioids, particularly with chronic administration, can manifest detectable neurotoxic activity in the absence of other toxic stimuli. The overt toxicity of morphine appears to be modest [37], but it should be noted that there is evidence that this μ-opioid, and others, can directly induce neurotoxic effects. Mu opioids including both morphine and fentanyl exert direct toxic effects on Purkinje cells in vitro, and fentanyl administration to rats in vivo induces damage to the limbic system and exacerbates cerebral ischemia in the forebrain [38–40]. Finally, μ-opioids exert a proapoptotic effect when combined at relatively low doses with various other apoptotic agents [41–45]. There is evidence of astrogliosis in heroin abusers [46], and the dopaminergic function of tyrosine hydroxylase terminals in the nucleus accumbens is inhibited [47]. Chronic morphine or heroin administration to rodents results in reduced striatal levels of synaptic dopamine and dopamine transporter [48–50]. These effects in the brain have been associated with the accumulation of perivascular infiltrates of macrophages and lymphocytes, suggesting that at least a part of the toxicity in these studies was the result of a low level of inflammatory activity in regions of brain tissue. Of course, other mechanisms are almost certainly involved in the manifestation of the gliosis just described, including the recent observation of hyperphosphorylated tau in the hippocampal neurons of heroin abusers [51, 52].
Opioids may promote toxic effects in the brain by inducing a pro-inflammatory response. This is somewhat counterintuitive, since opioid administration has been well documented to exert immunosuppressive activity (reviewed in [5]). However, an evaluation of published work from a number of investigators shows that opioid receptor activation can exert pleomorphic effects on the immune system, particularly with respect to the inflammatory response. Studies to determine the effects of morphine, and other μ-opioids, on the production of pro- and anti-inflammatory cytokines have produced conflicting results, and this is almost certainly due to the highly variable experimental systems employed for these studies (reviewed in [5, 36]). For example, morphine administration results in a reduction in the expression of interferon γ (IFNγ) and interleukin-2 (IL), cytokines that are critical for both acute inflammatory responses and adaptive immunity [53, 54]. Roy and her colleagues [55–57] working with both human blood leukocytes and murine splenocytes have shown that morphine polarizes toward a Th2 response, which would be expected to be less inflammatory. Sacerdote and her colleagues [58] have studied the effects of subcutaneous morphine administration on peritoneal macrophage function and reported a reduction in both baseline and lipopolysaccharide (LPS)-induced levels of IL-1β, tumor necrosis factor-α (TNFα), and IL-12. Recent work has also shown that morphine inhibits the expression of TNFα and IL-6 produced by human monocytes in response to bacterial peptidoglycan, but these effects required high concentrations of the opioid (10–100 μM) [59].
In contrast to these results, there are several reports which show that morphine, or other μ-opioids, induces the production of pro-inflammatory cytokines. Peng et al. [60] have reported an increase in the expression of both IL-12 and TNFα from murine peritoneal macrophages following morphine administration. These results are consistent with results with relatively low doses of morphine which show an increase in the expression of the pro-inflammatory cytokines IL-6 and TNFα, an effect which was due to the activation of the highly pro-inflammatory transcription factor NF-κB [61]. The latter results are particularly interesting in that the same investigation suggested that high morphine doses are inhibitory, suggesting that pharmacological doses of this drug may promote a more pro-inflammatory immune activity.
Morphine and other μ-opioids have been reported to upregulate NF-κB activity in neuronal cells. Treatment of rat cerebral cortex neurons with the MOR-selective agonist [D-ala2, N-Me-Phe4, Gly-ol5]enkephalin (DAMGO) induces NF-κB activation [62], and morphine treatment of the NT2-N neuronal cell line induced NF-κB promoter activity [63]. The activation of NF-κB has significant implications since it is critical for the expression of a large number of pro-inflammatory cytokines, including IL-1β, IL-6, and TNFα, and the chemokines CXCL8, CCL2, and CCL5 [64–70]. Both morphine [61] and the endogenous μ-opioids endomorphin 1 and endomorphin 2 [71] have been shown to upregulate NF-κB activity in monocyte/macrophage cell populations. More recently, our laboratory has reported the upregulation of NF-κB functional activity following administration of nanomolar concentrations of DAMGO to primary human peripheral blood leukocytes and MOR-transfected HEK293 cells [72]. Moreover, we found that the induction of NF-κB activity was essential for the opioid induction of the pro-inflammatory chemokine CCL2. Finally, the latter studies showed that the MOR-initiated signaling pathway for the induction of NF-κB is dependent on the activation of PKCζ, and treatment with a PKCζ-specific pseudosubstrate inhibitor blocks both the MOR-induced activation of NF-κB and the induction of CCL2 expression. Our studies have shown that the activation of MOR initiates a signaling pathway which results in the potent activation of PKCζ, and this atypical protein kinase C is involved in regulating multiple leukocyte functional activities [72, 73]. Previous studies have shown that PKCζ directly phosphorylates IKKβ, activating IKKβ, leading to the degradation of IκB [74, 75].
Morphine also induces the expression of TGFβ [76] in human peripheral blood leukocytes, and while this cytokine exhibits pleiotropic activities for the immune system, it is predominantly immunosuppressive. Given the immunosuppressive nature of this cytokine, it is possible that at least some of the reported negative effects of morphine on cytokine expression may be mediated by the production of TGFβ. We have found that TGFβ expression is induced following DAMGO administration to either human peripheral blood leukocytes or purified blood monocytes [77]. Our studies have shown that DAMGO also induces expression of both CCL5 and CXCR4 by human peripheral blood T cells and monocytes [78, 79], and we have recently shown that the upregulation of both CCL5 and CXCR4 is dependent on the initial production of TGFβ by these cell populations [77]. Moreover, these studies showed that both T cells and monocytes respond to TGFβ treatment by upregulating CCL5 and CXCR4 expression. These results are interesting in view of the anti-inflammatory effects of TGFβ, since this cytokine is necessary for the upregulation of a pro-inflammatory chemokine (CCL5) and a potentially pro-inflammatory chemokine receptor (CXCR4).
In addition to the potential pro-inflammatory effects of morphine, this opioid has been shown to promote neurodegeneration by weakening the integrity of the blood–brain barrier (BBB). Mahajan et al. [24], working with an in vitro BBB model, have shown that morphine inhibits the expression of the tight-junction zona occludin (ZO) proteins, ZO-1 and occludin, and increases the expression of junctional adhesion molecule (JAM)-1, leading to an increase in BBB permeability. These studies also showed that morphine induces an increase in the transmigration of peripheral blood leukocytes, suggesting the potential for increased traffic of inflammatory leukocytes into the brain with morphine administration. More recent studies have shown that morphine induces the expression of platelet-derived growth factor (PDGF) from brain microvascular endothelial cells in an in vitro model of the BBB [80]. This cytokine is a potent mitogen, exhibits chemoattractant activity, is highly pro-fibrotic, and has been reported to impair BBB integrity during ischemic stroke [81, 82]. The mechanism of BBB impairment mediated by PDGF is not clear, but it is known that PDGF is preferentially produced within the immune system by alternatively activated (M2) macrophages [83].
4 Neurodegeneration Mediated by Opiates in Association with HIV
A review of the effects of HIV infection, or the impact of HIV products, on the process of neurodegeneration is beyond the scope of this review. These issues will be discussed at length in other chapters of this book. However, the intersection between the neurodegenerative activity of opiates and HIV products will be discussed.
There is growing evidence that the combination of HIV infection and opiate drug abuse creates a heightened level of neurodegeneration compared with HIV or opiate use alone. Of course, mu opiates are well documented to alter the functional activity of neurons, microglia, astrocytes, neuronal precursors, and oligodendrocytes [84–99]. This is not altogether surprising, since each of these cell populations expresses MOR, albeit with diverse levels of expression in the various regions of the brain. However, the combination of HIV infection (and the release of HIV products into the brain milieu), with mu opiates, appears to target primarily the astrocytes and microglia and induce much greater pro-inflammatory and neurotoxic activity (reviewed in [100]).
4.1 Glial and Neuronal Cell Populations
Microglia play a critical role in HIV neuropathogenesis, and extensive activation of these cells (and infiltrating perivascular macrophages) is a common feature of the neurodegeneration associated with HIV infection [101, 102]. It is well known that the presence of activated macrophages and/or microglia correlates with the severity of the HIV-associated neurocognitive disorders (HAND); in fact, this correlation is stronger than the number of HIV-infected cells or viral load [103–106]. Both macrophages and microglia are subject to regulation mediated through opioid receptors, and these cells can exhibit substantial changes in functional activity with opiate administration. Experimental animal work has shown that systemic treatment with morphine induces an increase in the infiltration at sites of intrastriatal Tat injection [89]. These results are in agreement with published findings which show an association between increased numbers of microglia in the gray matter of the thalamus and hippocampus and encephalitis in opiate abusers [107]. Moreover, the accumulation of microglia expressing major histocompatibility complex type II (MHC II) and CD68 is increased in opiate abusers, when compared with non-abusers [108].
Astrocytes perform an essential set of functions in the development and maintenance of the brain and are important for the integrity of the BBB. Astrocytes are not susceptible to productive HIV infection (unlike macrophages and microglia), but these cells are important targets for the neurotoxic products of HIV. Astrocytes can exacerbate the neurodegenerative effects of HIV products by releasing mediators with potential toxicity such as nitric oxide, neurotransmitters, and pro-inflammatory cytokines, and collectively these serve to promote the HIV-mediated neuropathology. Of course, astrocytes also express opioid receptors, and morphine can augment, or accelerate, the neurotoxic activity of certain HIV products. For example, morphine administration together with HIV Tat can result in augmented cytokine and chemokine expression and, potentially, astrocyte death [90, 99]. Astrocytes are a major source of several of the chemokines within the brain, and opiate modulation of the expression of these mediators is an important aspect of the intersection between opioid receptors and the inflammatory response. The effects of opiates in combination with either HIV infection, or HIV products, will be discussed below.
The neurodegeneration observed in association with HIV infection leads to synaptodendritic injury which resembles the damage observed in other neurological diseases including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis [109–111]. The normal synaptodendritic network is characterized by highly complex and branching dendrites. However, in HIV encephalitis, the dendrites exhibit pruning, with dendritic beading, atrophy, and vacuolization [112]. Nevertheless, while this damage is typically sublethal, it is likely to contribute to the neurobehavioral deficits which are characteristic of HAND [113, 114]. At the same time, neuronal loss is a characteristic of HIV-associated neuropathology, and this is likely due to bystander effects mediated by reactive oxygen species and other neurotoxic products released from astrocytes and microglia [115–118]. Nevertheless, recent reports suggest that opiate abuse exacerbates the HIV-induced synaptodendritic damage and promotes the development of more severe neurobehavioral abnormalities [100, 119]. It should be pointed out that several reports have described the ability of MOR agonists to lessen the complexity of dendrites and diminish the density of dendritic spines [38, 120, 121].
Neurons appear to be directly susceptible to the HIV products Tat, gp120, and vpr, and the release of these products into the brain interstitium can result in neuronal damage [122–126]. Since neurons are not a source of productive HIV infection, the source of these products is primarily perivascular macrophages and microglial cell populations. This serves to highlight the fact that HIV-associated neurodegeneration is the product of glial transmission of neurotoxic products to the neuron [100]. Because the microglia express opioid receptors, these cells respond to treatment with exogenous opioid agonists and exhibit altered glial cell function. Recent reports suggest that opiate exposure reduces the level of HIV products which are required to trigger more pronounced neuropathology [87, 127]. Moreover, these studies show that morphine administration exacerbates the neurotoxicity of HIV Tat and gp120.
It is clear that μ-opioid agonists can also promote a neurodegenerative outcome through indirect effects mediated through changes in the levels of various neurotransmitters. For example, HIV Tat activates astrocytes leading to potent cytokine and inflammatory mediator release leading to restrictive glutamate uptake [90, 128]. HIV gp120 induces a similar astrocyte inflammatory response, and the combination of either of these HIV products and morphine enhances these responses, exacerbates the glutamate release, and reduces the glutamate excitotoxic threshold [129, 130]. In addition, several drugs of abuse, including the opiates, induce an increase in the levels of dopamine in the CNS. Morphine induces a twofold increase in levels of dopamine in the nucleus accumbens and caudate nucleus in rats [131]. Recent reports suggest that an elevated dopamine level, or administration of exogenous dopamine, alters monocyte/macrophage and T cell function [132–134]. Moreover, recent work suggests that dopamine administration promotes the replication of HIV in primary human macrophages [133]. In addition, the administration of either l-DOPA (the precursor of dopamine) or selegiline (inhibitor of dopamine catabolism) to SIV-infected macaques increases the SIV load in the brain [135, 136].
4.2 Inflammation and Immune Activation
As mentioned above, the capacity of MOR agonists to modulate the expression of pro-inflammatory cytokines has been the subject of a great deal of research (reviewed in [5]). The influence of opiates on chemokine expression is particularly important because these chemotactic cytokines are likely to be critical in the development of HIV-associated neurodegeneration. On the one hand, these factors are important for the traffic of infected monocytes across the BBB, and these cytokines may also promote the migration of infected T cells to lymph nodes to promote contact with noninfected target cells. The chemokines CCL2, CCL3, CCL4, CCL5, and CX3CL1 are chemokines that have been identified as contributors to the traffic of monocytes across the BBB, and CX3CL1 is particularly important for the migration of CD16+ monocytes that are highly susceptible to HIV infection [118, 137]. This relatively minor monocyte subpopulation that expresses both CD14 and CD16 has been reported to expand during HIV infection [138–143], and increased percentages of these cells correlate with HAD [144]. Monocytes and macrophages expressing high levels of CD16+ have been reported to be preferentially infected with HIV in brain tissue at autopsy [137]. In addition, the expression of the HIV co-receptor CCR5 is elevated on the CD16+ monocyte subset [145]. Recent analysis of monocyte subsets suggests that CD14+ CD16 cells exhibit a greater pro-inflammatory capacity, and the CD14+ 16+ cells possess strong responsiveness to viral pattern recognition epitopes and perform “patrolling” activity [146].
It is widely accepted that CCL2 is critically involved in directing the migration of infected monocytes across the BBB [147, 148]. With the accumulating inflammation in the underlying brain tissue, there is activation of the vascular endothelial cells in the BBB and secretion of IL-6 [149]. It is likely that the IL-6 produced at the BBB accelerates the transit of monocytes across the BBB. Both the virus infection and the accumulation of viral products induce elevated CCL2 expression from both the perivascular macrophages and astrocytes [150]. With greater accumulation of infected monocytes and macrophages, there is an expanding source of additional CCL2, resulting in biological amplification of the neuroinflammatory response.
As mentioned above, we have examined the effect of opiate administration on the expression of CCL2 by human peripheral blood leukocytes and purified monocytes [72, 77, 78]. In additional analysis, we also observed a significant induction of the chemokines CCL5 and CXCL10 following mu opiate treatment in vitro, and we suggest that the expression of these chemokines could promote the trafficking of noninfected target monocytes or T cells to the site of infected cell populations [78]. These results are consistent with more recent studies with mice which show that the combination of morphine and HIV Tat treatment of astrocytes upregulates the expression of the chemokines CCL2, CCL3, and CCL5 [88, 90]. These results suggest that the accumulation of Tat in the brain drives a pro-inflammatory chemokine response, and this response is accelerated by the administration of morphine. Additional studies showed that the morphine exacerbation of Tat-induced CCL2 expression is diminished in CCL5-knockout mice, suggesting that the regulation of CCL2 expression is mediated through a CCL5-dependent cooperative expression process [96]. It should be pointed out that these results are in contrast to a report showing downregulation of the expression of both CCL2 and CCL4 following morphine treatment of normal human astrocytes [151]. Additional work will be required to explain the divergent results in these studies.
Inflammation is a fundamental component of the neurodegenerative processes that are responsible for HIV-associated neurocognitive disorders. It is becoming apparent that elements of systemic inflammation are an important part of HIV pathogenesis, both in the CNS and the periphery [152–155]. Recent studies show that systemic inflammation promotes the development of HAND, and the augmentation of the pathology in the CNS can be independent of HIV replication [118, 147, 156]. For example, a recent analysis of a cohort of 922 HIV-infected subjects (the Study of Fat Redistribution and Metabolic Change in HIV infection [FRAM] study cohort) has shown that the pro-inflammatory biomarkers fibrinogen and C reactive protein (CRP) are significant and independent predictors of mortality [155]. Indeed, this study showed that these measures of inflammation retained predictive significance independent of circulating CD4 counts. A second study with the Strategies for Management of Antiretroviral Therapy trial showed a significant association for the inflammatory biomarkers IL-6, D-dimer, and CRP with mortality [157]. The persistent evidence of immune activation in these subjects, in the current highly active antiretroviral therapy (HAART) era, has been proposed as a significant contributor to disease progression [158]. The fact that chronic systemic inflammation is strongly associated with morbidity and mortality suggests that anti-inflammatory therapeutics may be beneficial as an adjunct to the standard ARV therapy currently in use.
The immune activation state that occurs with HIV infection and neurodegeneration is due in large part to the microbial translocation that is now believed to be common in these patients. Results from studies reported by Brenchley et al. [159] suggest that during HIV infection, a breakdown in the follicle-associated epithelium in the gut occurs, and this leads to translocation of gut flora through the gut wall, resulting in entry of microbial products into the bloodstream. This process is associated with depletion of leukocytes from the Peyer’s patches and a loss of lymphocytes from the lamina propria and mucosa-associated tissue (MALT). In healthy adults, approximately 80 % of the total lymphocytes of the body are contained within the MALT, and depletion of cells from these lymphoid structures can result in a substantial reduction of T cells following infection [160]. Recent analysis has shown that circulating LPS, LPS-binding protein, and sCD14 levels correlate significantly with progression of the disease, and monocytes obtained from these individuals exhibit a refractory response to LPS stimulation in vitro, suggesting that these cells had been stimulated in vivo with LPS [159, 161, 162]. It is now apparent that the toll-like receptor family is an important contributor to the persistent immune activation. This is not surprising given the entry of microbial components into the bloodstream following the gastrointestinal damage, but in addition, viral toll ligands also participate in the activation of TLR7 [163, 164]. Work with mice has demonstrated that sustained activation of TLR7 induces a state of chronic immune activation which resembles immune activation associated with HIV infection [165].
Studies reported by Hillburger et al. [166] have shown that mice treated with morphine using slow-release pellets develop bacterial sepsis as a likely result of microbial translocation. This study is particularly significant given the critical role for microbial translocation in the process of immune activation observed with HIV infection. However, this study did not examine the combined effect of morphine and HIV infection on the process of microbial translocation, and additional work on this issue would be valuable for our understanding of the influence of opiate use on immune status in HIV-infected patients.
The immune system is programmed to control the development of an inflammatory response, in part, through the production of immunosuppressive mediators such as IL-10 and TGFβ. Indeed, both TGFβ1 and IL-10 are upregulated in the CNS of patients who suffer with neurodegenerative diseases such as multiple sclerosis and Alzheimer’s disease, and TGFβ is upregulated in the CNS of patients with AIDS [167–170]. Analysis of brain tissue shows that TGFβ is readily detectable in macrophages, astrocytes, and microglial cells in the frontal cortex in patients with AIDS [170, 171]. The production of TGFβ in the CNS is considered anti-inflammatory and protective since there is evidence that it attenuates the level of astrocytosis which is characteristic of brain tissue in patients with HIV-associated dementia. In vitro analysis with astrocyte cultures shows that TGFβ inhibits cell proliferation and reduces glutamine synthetase [170]. Administration of TGFβ to microglial cell cultures results in downregulation of proliferation in response to either GM-CSF or M-CSF, and TGFβ inhibits the microglial expression of a number of pro-inflammatory cytokines and chemokines including IL-1, TNFα, CCL5, and CXCL8 [170, 171]. Moreover, TGFβ appears to inhibit expression of both complement factor 3 and inducible nitric oxide synthase, two potentially neurotoxic factors [172–175], and there are several reports which show that TGFβ inhibits microglial free radical production [176, 177]. Finally, TGFβ has been shown to inhibit HIV gp120-induced neuronal death, as well as calcium overloading, providing a degree of neuroprotection in HIV-infected brain tissue [178].
Based on reports from several investigators, the activation of MOR by opioid agonists appears to target TGFβ expression in leukocytes. For example, morphine treatment of human peripheral blood leukocytes downregulates the LPS- or PHA-induced expression of TNFα, and this effect is attenuated with the addition of anti-TGFβ antibodies [179]. In addition, morphine administration to human peripheral blood leukocytes upregulates TGFβ expression in response to either PHA or LPS [76]. More recently we observed that both human peripheral blood mononuclear cells and isolated peripheral blood monocytes upregulate TGFβ expression following activation of MOR [77]. It should be pointed out that TGFβ can exert pro-inflammatory activity in certain circumstances. For example, we have recently reported that mu opiates induce the expression of the pro-inflammatory chemokine CCL5, and the induction of expression is dependent on the initial expression of TGFβ [77]. In addition, TGFβ induces chemoattractant activity for monocytes and upregulates the expression of LFA-1 and the fibronectin receptor on monocytes [180–182]. These effects would be expected to promote adhesion of monocytes to endothelial cells and potentially promote traffic of monocytes across the BBB. Finally, there is evidence that TGFβ can induce monocyte expression of several pro-inflammatory cytokines [180, 183, 184]. While it is clear that the dominant role of TGFβ is to dampen the inflammatory response, it should be appreciated that the network of cytokines both in the periphery and in the brain can be quite complex, and TGFβ may exert a combination of effects as a part of a neurodegenerative disease process.
4.3 Interactions Between Opioid and Chemokine Receptors
It is well established that the chemokine receptors CCR5 and CXCR4 are the major HIV-1 co-receptors, and HIV strains can be distinguished based on the use of these co-receptors for target cell attachment and infection. The gp120 region of the HIV gp160 envelope protein possesses the capacity for binding to CD4, and one or more of the co-receptors, and this dictates cellular tropism for the virus. Virtually all HIV isolates from brain tissue use CCR5 rather than CXCR4 for viral attachment and are predominantly monocyte/macrophage tropic (R5 strains). Both of these chemokine receptors, and their chemokine ligands, are constitutively expressed in the brain.
Curiously, it appears that the expression of neuronal CXCR4 is upregulated, while the expression of CCR5 is reduced, in patients with HAND [185]. Both CXCR4 and CXCL12 are critical contributors to the development of the brain and play important roles in the maturation and maintenance of neuronal function in both the developing and adult brain [186, 187]. In the mature adult brain, CXCL12 has been shown to downregulate the expression of proapoptotic pathways and enhance neuronal survival [188, 189]. The role of CXCR4 in maintaining neuronal homeostasis is also mediated by modulation of the subunit composition of the NMDA receptor. Activation of CXCR4 leads to a reduction in the NR2B subunit of the NMDA receptor, and this substantially reduces excitotoxicity [190]. While X4 gp120 binds and activates CXCR4, the resulting signaling pathway is not identical to that which is induced by CXCL12 [191, 192]. Most notably, gp120 induces signaling elements which promote apoptosis and lead to a greater level of neuronal damage and cell death [193, 194].
Chronic morphine administration has recently been shown to inhibit the expression of the MAP kinases ERK1/2 and potentially attenuate the antiapoptotic activity of these kinases [195]. Moreover, morphine induces apoptosis in several regions of the brain, including both the frontal cortex and hippocampus [196]. Morphine has also been reported to significantly diminish dendritic spine complexity by reducing dendrite length and spine density [121, 197, 198]. Recent studies suggest that morphine mediates these effects on dendrite structure by attenuating the activity of the neurogenic differentiation 1 transcription factor (NeuroD), which is required for maintenance of dendritic spine stability [199]. As mentioned above, dendritic injury is a common feature of HAND, and evidence that morphine promotes dendritic simplification has significant implications.
The opioid receptors have the capacity to interact with the chemokine receptors that are expressed both within the CNS and in the periphery. We have reported studies which show that the activation of MOR leads to a significant upregulation of CCR5 and CXCR4 expression by human peripheral blood monocytes and T cell lymphoblasts [79]. This increase in the expression of CCR5 and CXCR4 was associated with an increase in susceptibility to infection with R5 and X4 strains of HIV-1, respectively [79]. These results are consistent with earlier work which has demonstrated that upregulation of co-receptor expression results in a corresponding increase in HIV replication [200, 201]. Treatment with morphine has also been reported to upregulate the expression of the chemokine receptors CCR2b, CCR3, and CCR5 by normal human astrocytes [151].
In addition to the capacity of the opiates to regulate the expression of chemokine receptors, there is considerable evidence that chemokine receptor function can be regulated through the action of opioid receptors. One of the mechanisms for the regulation of G protein-coupled receptor (GPCR) function is heterologous desensitization, a process in which the activation of one GPCR by its ligand results in the cross-inactivation of a second (unrelated) GPCR in the absence of the ligand for the second receptor (reviewed in [202]). Our laboratory and others have demonstrated that MOR can mediate cross-desensitization of several chemokine receptors, including CCR1, CCR2, CCR5, CXCR1, and CXCR2 [203, 204]. The biochemical basis for these interactions involves the sequential activation of multiple kinases, which leads to the activation of one or more members of the second messenger-dependent kinase family [202]. In the signaling pathway between MOR and CCR5, we have found that MOR activates PKCζ and this kinase phosphorylates and inactivates CCR5 within a period of less than 10 min [73]. The desensitization of CCR5 induced by MOR inhibits CCR5 function as measured by loss of chemotactic activity or a calcium mobilization response. In addition, HIV co-receptor function for cross-desensitized CCR5 is also lost when analyzed with R5 (but not X4) strains of HIV-1 [204].
Examination of the cross-talk between a number of GPCRs has led to the conclusion that there is a hierarchy which defines the interactions between these receptors [202]. In general, some GPCRs are strong cross-desensitizers but tend to be less sensitive as targets for the desensitization. On the other hand, certain GPCRs exhibit the opposite characteristics. For example, the formyl peptide receptor (FPR) is a relatively strong desensitizer, but this GPCR is difficult to cross-desensitize. Our laboratory has examined the interaction between MOR and CXCR4, and we have found that MOR is unable to cross-desensitize this receptor through this second messenger-dependent kinase pathway [204]. However, the Meucci laboratory [194] has described a cross-talk process in neuronal cells in which the activation of MOR results in inactivation of CXCR4, based on the loss of CXCR4 signaling activity. Their studies showed that the cross-desensitization induced through MOR resulted in the loss of the neuroprotective activity of CXCR4 in NMDA neurotoxicity studies. These results suggest that the cross-talk induced through activation of MOR would be very likely to contribute to the neurodegeneration associated with HIV infection.
We have reported results which show that the cross-desensitization between MOR and some susceptible chemokine receptors is bidirectional [205]. These studies show that MOR is cross-desensitized by CCR2, CCR5, CCR7, CX3CR1, and CXCR4, but not by CXCR1 or CXCR2. Moreover, the activation of CCR1, CCR5, or CXCR4 results in the loss of MOR-mediated analgesic activity in vivo [205–207]. These results suggest that in situations where the levels of pro-inflammatory chemokines are elevated in the brain, the threshold for sensation of pain is reduced. Clearly the neuroinflammation that is associated with HIV infection involves significantly increased levels of many pro-inflammatory chemokines. It is well known that heightened pain sensitivity (hyperalgesia) is associated with systemic inflammatory “flu-like” symptoms that include joint and muscle pain, fever, and somnolence [208, 209].
Finally, the results from our laboratory indicate that the MOR-induced cross-desensitization of CCR5 is apparent within 10–15 min and persists for at least 4–6 h. As mentioned above, this results in a loss of CCR5 co-receptor activity and a substantial reduction in susceptibility to R5 HIV infection [204]. The cross-desensitization of CCR5 can be prolonged if the MOR activation is sustained, but with acute opioid administration, the loss of co-receptor function is lost for the first several hours. This is followed by an increase in co-receptor function and increased R5 HIV susceptibility at 24–48 h [79], and this persists for several days. This suggests that the impact of opiates on R5 HIV susceptibility is likely to be complex, with confounding influences occurring at the level of co-receptor function.
5 Conclusion
The administration of opiates in the drug abuse population, in the context of HIV infection, promotes most of the neurodegenerative processes that take place as a part of the underlying viral infection. The brain would appear to be particularly susceptible to these effects because of the abundant number of cells which express MOR at relatively high levels. While opiates like morphine can exacerbate (or attenuate, depending on the conditions) systemic inflammatory processes, the data for the effects of these opiates would appear to be much less certain for the brain. The cells of the immune system in the periphery express much lower levels of the opioid receptors, and the impact of opiate administration is much more variable outside of the CNS. However, there are still many issues which need to be resolved in terms of the influence of opiate abuse on the development and progression of HIV-associated neurodegeneration. Of course, analysis of the progression of the disease in this organ is difficult because of the absence of tissue for longitudinal studies. Moreover, the drug-abusing population is very diverse, and controlled studies are extremely difficult because of the absence of subjects who do not abuse additional drugs. In fact, perhaps the most important questions that remain to be addressed will involve analysis of the effects of drug combinations, since this situation is much more relevant to the actual condition of patients. The most common drug combinations which should be studied are the combinations of opiates with tobacco or alcohol. The effects of these drug combinations on neurodegeneration in the context of HIV infection are almost entirely unknown.
References
Mathers BM, Degenhardt L, Ali H, Wiessing L, Hickman M, Mattick RP, et al. HIV prevention, treatment, and care services for people who inject drugs: a systematic review of global, regional, and national coverage. Lancet. 2010;375:1014–28.
Vlahov D, Robertson AM, Strathdee SA. Prevention of HIV infection among injection drug users in resource-limited settings. Clin Infect Dis. 2010;50 Suppl 3:S114–21.
Compton WM, Volkow ND. Abuse of prescription drugs and the risk of addiction. Drug Alcohol Depend. 2006;83 Suppl 1:S4–7.
Donahoe RM, Vlahov D. Opiates as potential cofactors in progression of HIV-1 infections to AIDS. J Neuroimmunol. 1998;83:77–87.
Finley MJ, Happel CM, Kaminsky DE, Rogers TJ. Opioid and nociceptin receptors regulate cytokine and cytokine receptor expression. Cell Immunol. 2008;252:146–54.
McCarthy L, Wetzel M, Sliker JK, Eisenstein TK, Rogers TJ. Opioids, opioid receptors, and the immune response. Drug Alcohol Depend. 2001;62:111–23.
Dutta R, Roy S. Mechanism(s) involved in opioid drug abuse modulation of HAND. Curr HIV Res. 2012;10:469–77.
Madera-Salcedo IK, Cruz SL, Gonzalez-Espinosa C. Morphine decreases early peritoneal innate immunity responses in Swiss-Webster and C57BL6/J mice through the inhibition of mast cell TNF- release. J Neuroimmunol. 2011;232:101–7.
Novick DM, Ochshorn M, Ghali V, Croxson TS, Mercer WD, Chiorazzi N, et al. Natural killer cell activity and lymphocyte subsets in parenteral heroin abusers and long-term methadone maintenance patients. J Pharmacol Exp Therapeut. 1989;250:606–10.
Kreek MJ, Khuri E, Flomenberg N, Albeck H, Ochshorn M. Immune status of unselected methadone maintained former heroin addicts. Progress Clin Biol Res. 1990;328:445–8.
Roy S, Ninkovic J, Banerjee S, Charboneau RG, Das S, Dutta R, et al. Opioid drug abuse and modulation of immune function: consequences in the susceptibility to opportunistic infections. J Neuroimmune Pharmacol. 2011;6:442–65.
Wang J, Barke RA, Charboneau R, Roy S. Morphine impairs host innate immune response and increases susceptibility to Streptococcus pneumoniae lung infection. J Immunol. 2005; 174:426–34.
MacFarlane AS, Peng X, Meissler Jr JJ, Rogers TJ, Geller EB, et al. Morphine increases susceptibility to oral Salmonella typhimurium infection. J Infect Dis. 2000;181:1350–8.
Tubaro E, Borelli G, Croce C, Cavallo G, Santiangeli C. Effect of morphine on resistance to infection. J Infect Dis. 1983;148:656–66.
Chao CC, Sharp BM, Pomeroy C, Filice GA, Peterson PK. Lethality of morphine in mice infected with Toxoplasma gondii. J Pharmacol Exp Therapeut. 1990;252:605–9.
Risdahl JM, Peterson PK, Chao CC, Pijoan C, Molitor TW. Effects of morphine dependence on the pathogenesis of swine herpesvirus infection. J Infect Dis. 1993;167:1281–7.
Starec M, Rouveix B, Sinet M, Chau F, Desforges B, Pocidalo JJ, et al. Immune status and survival of opiate- and cocaine-treated mice infected with Friend virus. J Pharmacol Exp Therapeut. 1991;259:745–50.
Wang J, Barke RA, Charboneau R, Schwendener R, Roy S. Morphine induces defects in early response of alveolar macrophages to Streptococcus pneumoniae by modulating TLR9-NF-kappa B signaling. J Immunol. 2008;180:3594–600.
Brack A, Rittner HL, Stein C. Immunosuppressive effects of opioids – clinical relevance. J Neuroimmune Pharmacol. 2011;6:490–502.
Donahoe RM, O’Neil SP, Marsteller FA, Novembre FJ, Anderson DC, Lankford-Turner P, et al. Probable deceleration of progression of Simian AIDS affected by opiate dependency: studies with a rhesus macaque/SIVsmm9 model. JAIDS. 2009;50:241–9.
Chuang RY, Chuang LF, Li Y, Kung HF, Killam Jr KF. SIV mutations detected in morphine-treated Macaca mulatta following SIVmac239 infection. Adv Exp Med Biol. 1995;373:175–81.
Marcario JK, Riazi M, Adany I, Kenjale H, Fleming K, Marquis J, et al. Effect of morphine on the neuropathogenesis of SIVmac infection in Indian Rhesus Macaques. J Neuroimmune Pharmacol. 2008;3:12–25.
Kumar R, Torres C, Yamamura Y, Rodriguez I, Martinez M, Staprans S, et al. Modulation by morphine of viral set point in rhesus macaques infected with simian immunodeficiency virus and simian-human immunodeficiency virus. J Virol. 2004;78:11425–8.
Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Fernandez SF, et al. Tight junction regulation by morphine and HIV-1 tat modulates blood-brain barrier permeability. J Clin Immunol. 2008;28:528–41.
Bokhari SM, Yao H, Bethel-Brown C, Fuwang P, Williams R, Dhillon NK, et al. Morphine enhances Tat-induced activation in murine microglia. J Neurovirol. 2009;15(3):219–28.
Lynch JL, Banks WA. Opiate modulation of IL-1alpha, IL-2, and TNF-alpha transport across the blood-brain barrier. Brain Behav Immun. 2008;22:1096–102.
Martinez AJ, Sell M, Mitrovics T, Stoltenburg-Didinger G, Iglesias-Rozas JR, Giraldo-Velasquez MA, et al. The neuropathology and epidemiology of AIDS. A Berlin experience. A review of 200 cases. Pathol Res Pract. 1995;191:427–43.
Bell JE, Donaldson YK, Lowrie S, McKenzie CA, Elton RA, Chiswick A, et al. Influence of risk group and zidovudine therapy on the development of HIV encephalitis and cognitive impairment in AIDS patients. Aids. 1996;10:493–9.
Platt DM, Grech DM, Rowlett JK, Spealman RD. Discriminative stimulus effects of morphine in squirrel monkeys: stimulants, opioids, and stimulant-opioid combinations. J Pharmacol Exp Therapeut. 1999;290:1092–100.
Stein C, Schafer M, Machelska H. Attacking pain at its source: new perspectives on opioids. Nat Med. 2003;9:1003–8.
Banerjee A, Strazza M, Wigdahl B, Pirrone V, Meucci O, Nonnemacher MR, et al. Role of mu-opioids as cofactors in human immunodeficiency virus type 1 disease progression and neuropathogenesis. J Neurovirol. 2011;17:291–302.
Belkowski SM, Zhu J, Liu-Chen LY, Eisenstein TK, Adler MW, et al. Sequence of kappa-opioid receptor cDNA in the R1.1 thymoma cell line. J Neuroimmunol. 1995;62:113–7.
Alicea C, Belkowski SM, Sliker JK, Zhu J, Liu-Chen LY, Eisenstein TK, et al. Characterization of kappa-opioid receptor transcripts expressed by T cells and macrophages. J Neuroimmunol. 1998;91:55–62.
Chuang TK, Killam Jr KF, Chuang LF, Kung HF, Sheng WS, et al. Mu opioid receptor gene expression in immune cells. Biochem Biophys Res Comm. 1995;216:922–30.
Bohn LM, Belcheva MM, Coscia CJ. Mu-opioid agonist inhibition of kappa-opioid receptor-stimulated extracellular signal-regulated kinase phosphorylation is dynamin-dependent in C6 glioma cells. J Neurochem. 2000;74:574–81.
Rogers TJ, Peterson PK. Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol. 2003;24:116–21.
Rogers RD, Everitt BJ, Baldacchino A, Blackshaw AJ, Swainson R, Wynne K, et al. Dissociable deficits in the decision-making cognition of chronic amphetamine abusers, opiate abusers, patients with focal damage to prefrontal cortex, and tryptophan-depleted normal volunteers: evidence for monoaminergic mechanisms. Neuropsychopharmacology. 1999;20:322–39.
Hauser KF, Gurwell JA, Turbek CS. Morphine inhibits Purkinje cell survival and dendritic differentiation in organotypic cultures of the mouse cerebellum. Exp Neurol. 1994;130: 95–105.
Kofke WA, Garman RH, Stiller RL, Rose ME, Garman R. Opioid neurotoxicity: fentanyl dose-response effects in rats. Anesthesia Analgesia. 1996;83:1298–306.
Kofke WA, Garman RH, Garman R, Rose ME. Opioid neurotoxicity: fentanyl-induced exacerbation of cerebral ischemia in rats. Brain Res. 1999;818:326–34.
Singhal PC, Sharma P, Kapasi AA, Reddy K, Franki N, Gibbons N. Morphine enhances macrophage apoptosis. J Immunol. 1998;160:1886–93.
Nair MP, Schwartz SA, Polasani R, Hou J, Sweet A, Chadha KC. Immunoregulatory effects of morphine on human lymphocytes. Clin Diagn Lab Immunol. 1997;4:127–32.
Yin DL, Ren XH, Zheng ZL, Pu L, Jiang LZ, Ma L, et al. Etorphine inhibits cell growth and induces apoptosis in SK-N-SH cells: involvement of pertussis toxin-sensitive G proteins. Neurosci Res Suppl. 1997;29:121–7.
Singhal PC, Reddy K, Franki N, Sanwal V, Gibbons N. Morphine induces splenocyte apoptosis and enhanced mRNA expression of cathepsin-B. Inflammation. 1997;21:609–17.
Singhal PC, Kapasi AA, Reddy K, Franki N, Gibbons N, Ding G. Morphine promotes apoptosis in Jurkat cells. J Leuk Biol. 1999;66:650–8.
Buttner A. Review: the neuropathology of drug abuse. Neuropathol Appl Neurobiol. 2011;37: 118–34.
Kish SJ, Kalasinsky KS, Derkach P, Schmunk GA, Guttman M, Ang L, et al. Striatal dopaminergic and serotonergic markers in human heroin users. Neuropsychopharmacology. 2001;24: 561–7.
Acquas E, Carboni E, Di CG. Profound depression of mesolimbic dopamine release after morphine withdrawal in dependent rats. Eur J Pharmacol. 1991;193:133–4.
Crippens D, Robinson TE. Withdrawal from morphine or amphetamine: different effects on dopamine in the ventral-medial striatum studied with microdialysis. Brain Res. 1994;650:56–62.
Simantov R. Chronic morphine alters dopamine transporter density in the rat brain: possible role in the mechanism of drug addiction. Neurosci Lett. 1993;163:121–4.
Anthony IC, Norrby KE, Dingwall T, Carnie FW, Millar T, Arango JC, et al. Predisposition to accelerated Alzheimer-related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain. 2010;133:12–98.
Ramage SN, Anthony IC, Carnie FW, Busuttil A, Robertson R, Bell JE, et al. Hyperphosphorylated tau and amyloid precursor protein deposition is increased in the brains of young drug abusers. Neuropathol Appl Neurobiol. 2005;31:439–48.
Peterson PK, Sharp B, Gekker G, Brummitt C, Keane WF. Opioid-mediated suppression of interferon-gamma production by cultured peripheral blood mononuclear cells. J Clin Invest. 1987;80:824–31.
Lysle DT, Coussons ME, Watts VJ, Bennett EH, Dykstra LA. Morphine-induced alterations of immune status: dose dependency, compartment specificity and antagonism by naltrexone. J Pharmacol Exp Therapeut. 1993;265:1071–8.
Roy S, Balasubramanian S, Sumandeep S, Charboneau R, Wang J, Melnyk D, et al. Morphine directs T cells toward T(H2) differentiation. Surgery. 2001;130:304–9.
Roy S, Wang J, Gupta S, Charboneau R, Loh HH, Barke RA. Chronic morphine treatment differentiates T helper cells to Th2 effector cells by modulating transcription factors GATA 3 and T-bet. J Neuroimmunol. 2004;147:78–81.
Roy S, Wang J, Kelschenbach J, Koodie L, Martin J. Modulation of immune function by morphine: implications for susceptibility to infection. J Neuroimmune Pharmacol. 2006;1:77–89.
Martucci C, Franchi S, Lattuada D, Panerai AE, Sacerdote P. Differential involvement of RelB in morphine-induced modulation of chemotaxis, NO, and cytokine production in murine macrophages and lymphocytes. J Leuk Biol. 2007;81:344–54.
Bonnet MP, Beloeil H, Benhamou D, Mazoit JX, Asehnoune K. The mu opioid receptor mediates morphine-induced tumor necrosis factor and interleukin-6 inhibition in toll-like receptor 2-stimulated monocytes. Anesthesia Analgesia. 2008;106:1142–9.
Peng X, Mosser DM, Adler MW, Rogers TJ, Meissler Jr JJ, Eisenstein TK. Morphine enhances interleukin-12 and the production of other pro-inflammatory cytokines in mouse peritoneal macrophages. J Leuk Biol. 2000;68:723–8.
Roy S, Cain KJ, Chapin RB, Charboneau RG, Barke RA. Morphine modulates NF kappa B activation in macrophages. Biochem Biophys Res Comm. 1998;245:392–6.
Hou YN, Vlaskovska M, Cebers G, Kasakov L, Liljequist S, Terenius L, et al. A mu-receptor opioid agonist induces AP-1 and NF-kappa B transcription factor activity in primary cultures of rat cortical neurons. Neurosci Lett. 1996;212:159–62.
Wang X, Douglas SD, Commons KG, Pleasure DE, Lai J, Ho C, et al. A non-peptide substance P antagonist (CP-96,345) inhibits morphine-induced NF-kappa B promoter activation in human NT2-N neurons. J Neurosci Res. 2004;75:544–53.
Kuprash DV, Udalova IA, Turetskaya RL, Rice NR, Nedospasov SA. Conserved kappa B element located downstream of the tumor necrosis factor alpha gene: distinct NF-kappa B binding pattern and enhancer activity in LPS activated murine macrophages. Oncogene. 1995;11:97–106.
Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur J Immunol. 1997;27:1091–7.
Moriuchi H, Moriuchi M, Fauci AS. Nuclear factor-kappa B potently up-regulates the promoter activity of RANTES, a chemokine that blocks HIV infection. J Immunol. 1997;158: 3483–91.
Mukaida N, Okamoto S, Ishikawa Y, Matsushima K. Molecular mechanism of interleukin-8 gene expression. J Leuk Biol. 1994;56:554–8.
Stein B, Baldwin Jr AS. Distinct mechanisms for regulation of the interleukin-8 gene involve synergism and cooperativity between C/EBP and NF-kappa B. Mol Cell Biol. 1993;13:7191–8.
Hiscott J, Marois J, Garoufalis J, D’Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13:6231–40.
Galien R, Evans HF, Garcia T. Involvement of CCAAT/enhancer-binding protein and nuclear factor-kappa B binding sites in interleukin-6 promoter inhibition by estrogens. Mol Endocrinol. 1996;10:713–22.
Azuma Y, Ohura K. Endomorphins 1 and 2 inhibit IL-10 and IL-12 production and innate immune functions, and potentiate NF-kappaB DNA binding in THP-1 differentiated to macrophage-like cells. Scand J Immunol. 2002;56:260–9.
Happel C, Kutzler M, Rogers TJ. Opioid-induced chemokine expression requires NF-kB activity: the role of PKC. J Leuk Biol. 2011;89:301–9.
Song C, Rahim RT, Davey PC, Bednar F, Bardi G, Zhang L, et al. Protein kinase Czeta mediates μ-opioid receptor-induced cross-desensitization of chemokine receptor CCR5. J Biol Chem. 2011;286:20354–65.
Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401.
Law PY, Loh HH, Wei LN. Insights into the receptor transcription and signaling: implications in opioid tolerance and dependence. Neuropharmacology. 2004;47 Suppl 1:300–11.
Chao CC, Hu S, Molitor TW, Zhou Y, Murtaugh MP, Tsang M, et al. Morphine potentiates transforming growth factor-beta release from human peripheral blood mononuclear cell cultures. J Pharmacol Exp Therapeut. 1992;262:19–24.
Happel C, Steele AD, Finley MJ, Kutzler MA, Rogers TJ. DAMGO-induced expression of chemokines and chemokine receptors: the role of TGF-β1. J Leukoc Biol. 2008;83:956–63.
Wetzel MA, Steele AD, Eisenstein TK, Adler MW, Henderson EE, Rogers TJ. Mu-opioid induction of monocyte chemoattractant protein-1, RANTES, and IFN-gamma-inducible protein-10 expression in human peripheral blood mononuclear cells. J Immunol. 2000;165:6519–24.
Steele AD, Henderson EE, Rogers TJ. Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology. 2003;309:99–107.
Wen H, Lu Y, Yao H, Buch S, Wen H, Lu Y, et al. Morphine induces expression of platelet-derived growth factor in human brain microvascular endothelial cells: implication for vascular permeability. PLoS One. 2011;6:e21707.
Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–73.
Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–7.
Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000;204:19–28.
Gupta S, Knight AG, Gupta S, Knapp PE, Hauser KF, Keller JN, et al. HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci Lett. 2010;485:233–6.
Turchan-Cholewo J, Liu Y, Gartner S, Reid R, Jie C, Peng X, et al. Increased vulnerability of ApoE4 neurons to HIV proteins and opiates: protection by diosgenin and L-deprenyl. Neurobiol Dis. 2006;23:109–19.
Malik S, Khalique H, Buch S, Seth P, Malik S, Khalique H, et al. A growth factor attenuates HIV-1 Tat and morphine induced damage to human neurons: implication in HIV/AIDS-drug abuse cases. PLoS One. 2011;6:e18116.
Hu S, Sheng WS, Lokensgard JR, Peterson PK. Morphine potentiates HIV-1 gp120-induced neuronal apoptosis. J Infect Dis. 2005;191:886–9.
El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, et al. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-kappaB trafficking and transcription. PLoS One. 2008;3:e4093.
El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, et al. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–46.
El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106.
Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–59.
Gurwell JA, Duncan MJ, Maderspach K, Stiene-Martin A, Elde RP, Hauser KF. kappa-opioid receptor expression defines a phenotypically distinct subpopulation of astroglia: relationship to Ca2+ mobilization, development, and the antiproliferative effect of opioids. Brain Res. 1996;737:175–87.
Hauser KF, Stiene-Martin A, Mattson MP, Elde RP, Ryan SE, Godleske CC. mu-Opioid receptor-induced Ca2+ mobilization and astroglial development: morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca(2+)-dependent mechanism. Brain Res. 1996;720:191–203.
Stiene-Martin A, Mattson MP, Hauser KF. Opiates selectively increase intracellular calcium in developing type-1 astrocytes: role of calcium in morphine-induced morphologic differentiation. Brain Res Dev Brain Res. 1993;76:189–96.
Turchan-Cholewo J, Dimayuga FO, Ding Q, Keller JN, Hauser KF, Knapp PE, et al. Cell-specific actions of HIV-Tat and morphine on opioid receptor expression in glia. J Neurosci Res. 2008;86:2100–10.
El-Hage N, Bruce-Keller AJ, Knapp PE, Hauser KF. CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat-exposed mice. J Neuroimmune Pharmacol. 2008;3:275–85.
Hauser KF, Hahn YK, Adjan VV, Zou S, Buch SK, Nath A, et al. HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia. 2009;57:194–206.
Buch SK, Khurdayan VK, Lutz SE, Knapp PE, El-Hage N, Hauser KF. Glial-restricted precursors: patterns of expression of opioid receptors and relationship to human immunodeficiency virus-1 Tat and morphine susceptibility in vitro. Neuroscience. 2007;146:1546–54.
Khurdayan VK, Buch S, El-Hage N, Lutz SE, Goebel SM, Singh IN, et al. Preferential vulnerability of astroglia and glial precursors to combined opioid and HIV-1 Tat exposure in vitro. Eur J Neurosci. 2004;19:3171–82.
Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE. Opiate drug use and the pathophysiology of neuroAIDS. Curr HIV Res. 2012;10:435–52.
Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–94.
Persidsky Y, Gendelman HE. Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J Leuk Biol. 2003;74:691–701.
Tyor WR, Wesselingh SL, Griffin JW, McArthur JC, Griffin DE. Unifying hypothesis for the pathogenesis of HIV-associated dementia complex, vacuolar myelopathy, and sensory neuropathy. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:379–88.
Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755–62.
Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81.
Adle-Biassette H, Chretien F, Wingertsmann L, Hery C, Ereau T, Scaravilli F, et al. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–33.
Arango JC, Simmonds P, Brettle RP, Bell JE. Does drug abuse influence the microglial response in AIDS and HIV encephalitis? Aids. 2004;18 Suppl 1:S69–74.
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Does drug abuse alter microglial phenotype and cell turnover in the context of advancing HIV infection? Neuropathol Appl Neurobiol. 2005;31:325–38.
Masliah E. Mechanisms of synaptic pathology in Alzheimer’s disease. J Neural Transm Suppl. 1998;53:147–58.
Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett. 1994;174:67–72.
Law AJ, Weickert CS, Hyde TM, Kleinman JE, Harrison PJ. Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am J Psychiatr. 2004;161:1848–55.
Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–72.
Everall IP, Everall IP. Neuronal damage - recent issues and implications for therapy. J Neurovirol. 2000;6 Suppl 1:S103–5.
Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, et al. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9: 209–17.
Agrawal L, Louboutin JP, Marusich E, Reyes BA, Van Bockstaele EJ, Strayer DS. Dopaminergic neurotoxicity of HIV-1 gp120: reactive oxygen species as signaling intermediates. Brain Res. 2010;1306:116–30.
Gray F, Adle-Biassette H, Brion F, Ereau T, le Maner I, Levy V, et al. Neuronal apoptosis in human immunodeficiency virus infection. J Neurovirol. 2000;6 Suppl 1:S38–43.
Gray F, Adle-Biassette H, Chretien F, Lorin dG, Force G, Keohane C. Neuropathology and neurodegeneration in human immunodeficiency virus infection. Pathogenesis of HIV-induced lesions of the brain, correlations with HIV-associated disorders and modifications according to treatments. Clin Neuropathol. 2001;20:146–55.
Grovit-Ferbas K, Harris-White ME. Thinking about HIV: the intersection of virus, neuroinflammation and cognitive dysfunction. Immunologic Res. 2010;48:40–58.
Fitting S, Xu R, Bull C, Buch SK, El-Hage N, Nath A, et al. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol. 2010;177:1397–410.
Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47 Suppl 1:33–46.
Robinson TE, Kolb B. Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse. 1999;33:160–2.
Nath A. Pathobiology of human immunodeficiency virus dementia. Semin Neurol. 1999;19:113–27.
Dreyer EB, Kaiser PK, Offermann JT, Lipton SA. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990;248:364–7.
Haughey NJ, Holden CP, Nath A, Geiger JD. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem. 1999;73:1363–74.
Piller SC, Jans P, Gage PW, Jans DA. Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc Natl Acad Sci U S A. 1998;95:4595–600.
Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12 Suppl 1:893–904 [Review] [173 refs].
Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, et al. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience. 2001;102:555–63.
Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, et al. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at u-opioid receptor-expressing glia. Brain. 2011;134:12–31.
Podhaizer EM, Zou S, Fitting S, Samano KL, El-Hage N, Knapp PE, et al. Morphine and gp120 toxic interactions in striatal neurons are dependent on HIV-1 strain. J Neuroimmune Pharmacol. 2012;7:877–91.
Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–8.
Di CG, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–8.
Levite M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr Opin Pharmacol. 2008;8:460–71.
Gaskill PJ, Calderon TM, Luers AJ, Eugenin EA, Javitch JA, Berman JW. Human immunodeficiency virus (HIV) infection of human macrophages is increased by dopamine: a bridge between HIV-associated neurologic disorders and drug abuse. Am J Pathol. 2009;175: 1148–59.
Gaskill PJ, Carvallo L, Eugenin EA, Berman JW. Characterization and function of the human macrophage dopaminergic system: implications for CNS disease and drug abuse. J Neuroinflammation. 2012;9:203.
Czub S, Koutsilieri E, Sopper S, Czub M, Stahl-Hennig C, Muller JG, et al. Enhancement of central nervous system pathology in early simian immunodeficiency virus infection by dopaminergic drugs. Acta Neuropathol. 2001;101:85–91.
Czub S, Czub M, Koutsilieri E, Sopper S, Villinger F, Muller JG, et al. Modulation of simian immunodeficiency virus neuropathology by dopaminergic drugs. Acta Neuropathol. 2004;107:216–26.
Dunfee R, Thomas ER, Gorry PR, Wang J, Ancuta P, Gabuzda D, et al. Mechanisms of HIV-1 neurotropism. Curr HIV Res. 2006;4:267–78.
Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N. CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995;25:3418–24.
Allen JB, Wong HL, Guyre PM, Simon GL, Wahl SM. Association of circulating receptor Fc gamma RIII-positive monocytes in AIDS patients with elevated levels of transforming growth factor-beta. J Clin Invest. 1991;87:1773–9.
Locher C, Vanham G, Kestens L, Kruger M, Ceuppens JL, Vingerhoets J, et al. Expression patterns of Fc gamma receptors, HLA-DR and selected adhesion molecules on monocytes from normal and HIV-infected individuals. Clin Exp Immunol. 1994;98:115–22.
Nockher WA, Bergmann L, Scherberich JE. Increased soluble CD14 serum levels and altered CD14 expression of peripheral blood monocytes in HIV-infected patients. Clin Exp Immunol. 1994;98:369–74.
Dunne J, Feighery C, Whelan A. Beta-2-microglobulin, neopterin and monocyte Fc gamma receptors in opportunistic infections of HIV-positive patients. Br J Biomed Sci. 1996;53:263–9.
Ancuta P, Weiss L, Haeffner-Cavaillon N. CD14 + CD16++ cells derived in vitro from peripheral blood monocytes exhibit phenotypic and functional dendritic cell-like characteristics. Eur J Immunol. 2000;30:1872–83.
Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS. Unique monocyte subset in patients with AIDS dementia. Lancet. 1997;349:692–5.
Weber C, Belge KU, von Hundelshausen P, Draude G, Steppich B, Mack M, et al. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leuk Biol. 2000;67:699–704.
Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ, Geissmann F, et al. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10:453–60.
Kraft-Terry SD, Buch SJ, Fox HS, Gendelman HE. A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron. 2009;64:133–45.
Peng F, Dhillon NK, Yao H, Zhu X, Williams R, Buch S, et al. Mechanisms of platelet-derived growth factor-mediated neuroprotection – implications in HIV dementia. Eur J Neurosci. 2008;28:1255–64.
Chaudhuri A, Yang B, Gendelman HE, Persidsky Y, Kanmogne GD. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood. 2008;111:2062–72.
Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–106.
Mahajan SD, Schwartz SA, Aalinkeel R, Chawda RP, Sykes DE, Nair MP. Morphine modulates chemokine gene regulation in normal human astrocytes. Clin Immunol. 2005;115:323–32.
Deeks SG. Immune dysfunction, inflammation, and accelerated aging in patients on antiretroviral therapy. Top HIV Med. 2009;17:118–23.
Neuhaus J, Jacobs Jr DR, Baker JV, Calmy A, Duprez D, La RA, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. 2010;201:1788–95.
Reingold J, Wanke C, Kotler D, Lewis C, Tracy R, Heymsfield S, et al. Association of HIV infection and HIV/HCV coinfection with C-reactive protein levels: the fat redistribution and metabolic change in HIV infection (FRAM) study. JAIDS. 2008;48:142–8.
Tien PC, Choi AI, Zolopa AR, Benson C, Tracy R, Scherzer R, et al. Inflammation and mortality in HIV-infected adults: analysis of the FRAM study cohort. JAIDS. 2010;55: 316–22.
Kaul M, Lipton SA. Mechanisms of neuroimmunity and neurodegeneration associated with HIV-1 infection and AIDS. J Neuroimmune Pharmacol. 2006;1:138–51.
Kuller LH, Tracy R, Belloso W, De WS, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203.
Gannon P, Khan MZ, Kolson DL. Current understanding of HIV-associated neurocognitive disorders pathogenesis. Curr Opin Neurol. 2011;24:275–83.
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71.
Wallet MA, Rodriguez CA, Yin L, Saporta S, Chinratanapisit S, Hou W, et al. Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T-cell activation following therapy. Aids. 2010;24:1281–90.
Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199:1177–85.
Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–9.
Meier A, Alter G, Frahm N, Sidhu H, Li B, Bagchi A, et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J Virol. 2007;81:8180–91.
Beignon AS, McKenna K, Skoberne M, Manches O, Dasilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–75.
Baenziger S, Heikenwalder M, Johansen P, Schlaepfer E, Hofer U, Miller RC, et al. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood. 2009;113:377–88.
Hilburger ME, Adler MW, Truant AL, Meissler Jr JJ, Satishchandran V, Rogers TJ, et al. Morphine induces sepsis in mice. J Infect Dis. 1997;176:183–8.
Peress NS, Perillo E, Seidman RJ. Glial transforming growth factor (TGF)-beta isotypes in multiple sclerosis: differential glial expression of TGF-beta 1, 2 and 3 isotypes in multiple sclerosis. J Neuroimmunol. 1996;71:115–23.
Carrieri PB, Provitera V, De RT, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. 1998;20:373–82.
Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–8.
Vitkovic L, Maeda S, Sternberg E. Anti-inflammatory cytokines: expression and action in the brain. [Review] [205 refs]. Neuroimmunomodulation. 2001;9:295–312.
Benveniste EN. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998;9:259–75.
Barnum SR, Jones JL. Transforming growth factor-beta 1 inhibits inflammatory cytokine-induced C3 gene expression in astrocytes. J Immunol. 1994;152:765–73.
Vodovotz Y, Geiser AG, Chesler L, Letterio JJ, Campbell A, Lucia MS, et al. Spontaneously increased production of nitric oxide and aberrant expression of the inducible nitric oxide synthase in vivo in the transforming growth factor beta 1 null mouse. J Exp Med. 1996;183:2337–42.
Park SK, Grzybicki D, Lin HL, Murphy S. Modulation of inducible nitric oxide synthase expression in astroglial cells. Neuropharmacology. 1994;33:1419–23.
Bottner M, Krieglstein K, Unsicker K. The transforming growth factor-betas: structure, signaling, and roles in nervous system development and functions. J Neurochem. 2000;75: 2227–40.
Chao CC, Hu S, Peterson PK. Modulation of human microglial cell superoxide production by cytokines. J Leuk Biol. 1995;58:65–70.
Hu S, Sheng WS, Peterson PK, Chao CC. Cytokine modulation of murine microglial cell superoxide production. Glia. 1995;13:45–50.
Meucci O, Miller RJ. gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGF-beta1. J Neurosci. 1996;16:4080–8.
Chao CC, Molitor TW, Close K, Hu S, Peterson PK. Morphine inhibits the release of tumor necrosis factor in human peripheral blood mononuclear cell cultures. Int J Immunopharmacol. 1993;15:447–53.
Wahl SM, Hunt DA, Wakefield LM, Cartney-Francis N, Wahl LM, Roberts AB, et al. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. 1987;84:5788–92.
Wiseman DM, Polverini PJ, Kamp DW, Leibovich SJ. Transforming growth factor-beta (TGF beta) is chemotactic for human monocytes and induces their expression of angiogenic activity. Biochem Biophys Res Comm. 1988;157:793–800.
Wahl SM, Allen JB, Weeks BS, Wong HL, Klotman PE. Transforming growth factor beta enhances integrin expression and type IV collagenase secretion in human monocytes. Proc Natl Acad Sci U S A. 1993;90:4577–81.
Riddick CA, Serio KJ, Hodulik CR, Ring WL, Regan MS, Bigby TD. TGF-beta increases leukotriene C4 synthase expression in the monocyte-like cell line, THP-1. J Immunol. 1999;162:1101–7.
Turner M, Chantry D, Feldmann M. Transforming growth factor beta induces the production of interleukin 6 by human peripheral blood mononuclear cells. Cytokine. 1990;2:211–6.
Petito CK, Roberts B, Cantando JD, Rabinstein A, Duncan R. Hippocampal injury and alterations in neuronal chemokine co-receptor expression in patients with AIDS. J Neuropathol Exp Neurol. 2001;60:377–85.
Li M, Ransohoff RM. Multiple roles of chemokine CXCL12 in the central nervous system: a migration from immunology to neurobiology. Prog Neurobiol. 2008;84:116–31.
Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Role of the alpha-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia. 2003;42:139–48.
Khan MZ, Brandimarti R, Shimizu S, Nicolai J, Crowe E, Meucci O. The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell Death Differ. 2008;15:1663–72.
Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21.
Nicolai J, Burbassi S, Rubin J, Meucci O. CXCL12 inhibits expression of the NMDA receptor’s NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Disease. 2010;1:e33.
Khan MZ, Brandimarti R, Patel JP, Huynh N, Wang J, Huang Z, et al. Apoptotic and antiapoptotic effects of CXCR4: is it a matter of intrinsic efficacy? Implications for HIV neuropathogenesis. AIDS Res Hum Retrovirus. 2004;20:1063–71.
Toth PT, Ren D, Miller RJ. Regulation of CXCR4 receptor dimerization by the chemokine SDF-1alpha and the HIV-1 coat protein gp120: a fluorescence resonance energy transfer (FRET) study. J Pharmacol Exp Therapeut. 2004;310:8–17.
Festa L, Meucci O. Effects of opiates and HIV proteins on neurons: the role of ferritin heavy chain and a potential for synergism. Curr HIV Res. 2012;10:453–62.
Patel JP, Sengupta R, Bardi G, Khan MZ, Mullen-Przeworski A, Meucci O. Modulation of neuronal CXCR4 by the micro-opioid agonist DAMGO. J Neurovirol. 2006;12:492–500.
Ferrer-Alcon M, Garcia-Fuster MJ, La HR, Garcia-Sevilla JA. Long-term regulation of signalling components of adenylyl cyclase and mitogen-activated protein kinase in the pre-frontal cortex of human opiate addicts. J Neurochem. 2004;90:220–30.
Atici S, Cinel L, Cinel I, Doruk N, Aktekin M, Akca A, et al. Opioid neurotoxicity: comparison of morphine and tramadol in an experimental rat model. Int J Neurosci. 2004;114:1001–11.
Liao D, Lin H, Law PY, Loh HH. Mu-opioid receptors modulate the stability of dendritic spines. Proc Natl Acad Sci U S A. 2005;102:1725–30.
Li Y, Wang H, Niu L, Zhou Y. Chronic morphine exposure alters the dendritic morphology of pyramidal neurons in visual cortex of rats. Neurosci Lett. 2007;418:227–31.
Zheng H, Zeng Y, Chu J, Kam AY, Loh HH, Law PY. Modulations of NeuroD activity contribute to the differential effects of morphine and fentanyl on dendritic spine stability. J Neurosci. 2010;30:8102–10.
Tuttle DL, Harrison JK, Anders C, Sleasman JW, Goodenow MM. Expression of CCR5 increases during monocyte differentiation and directly mediates macrophage susceptibility to infection by human immunodeficiency virus type 1. J Virol. 1998;72:4962–9.
Secchiero P, Zella D, Capitani S, Gallo RC, Zauli G. Extracellular HIV-1 tat protein up-regulates the expression of surface CXC-chemokine receptor 4 in resting CD4+ T cells. J Immunol. 1999;162:2427–31.
Steele AD, Szabo I, Bednar F, Rogers TJ. Interactions between opioid and chemokine receptors: heterologous desensitization. Cytokine Growth Factor Rev. 2002;13:209–22.
Grimm MC, Ben Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, et al. Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J Exp Med. 1998;188:317–25.
Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, et al. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leuk Biol. 2003;74:1074–82.
Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, et al. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci U S A. 2002;99:10276–81.
Chen X, Geller EB, Rogers TJ, Adler MW. The chemokine CX3CL1/fractalkine interferes with the antinociceptive effect induced by opioid agonists in the periaqueductal grey of rats. Brain Res. 2007;1153:52–7.
Chen X, Geller EB, Rogers TJ, Adler MW, Chen X, Geller EB, et al. Rapid heterologous desensitization of antinociceptive activity between mu or delta opioid receptors and chemokine receptors in rats. Drug Alcohol Depend. 2007;88:36–41.
Watkins LR, Maier SF, Goehler LE. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995;63:289–302.
Junger H, Sorkin LS. Nociceptive and inflammatory effects of subcutaneous TNFalpha. Pain. 2000;85:145–51.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Rogers, T.J. (2014). Drugs of Abuse and NeuroAIDS: Opiates. In: Peterson, P., Toborek, M. (eds) Neuroinflammation and Neurodegeneration. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1071-7_22
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1071-7_22
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1070-0
Online ISBN: 978-1-4939-1071-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)