
Abstract
Despite effective and widely available suppressive anti-HIV therapy, the prevalence of mild neurocognitive dysfunction continues to increase. HIV-associated neurocognitive disorder (HAND) is a multifactorial disease with sustained central nervous system inflammation and immune activation as prominent features. Inflammatory macrophages, HIV-infected and uninfected, play a central role in the development of HIV dementia. There is a critical need to identify biomarkers and to better understand the molecular mechanisms leading to cognitive dysfunction in HAND. In this regard, we identified through a subtractive hybridization strategy osteopontin (OPN, SPP1, gene) an inflammatory marker, as an upregulated gene in HIV-infected primary human monocyte-derived macrophages. Knockdown of OPN in primary macrophages resulted in a threefold decrease in HIV-1 replication. Ectopic expression of OPN in the TZM-bl cell line significantly enhanced HIV infectivity and replication. A significant increase in the degradation of the NF-κB inhibitor, IκBα and an increase in the nuclear-to-cytoplasmic ratio of NF-κB were found in HIV-infected cells expressing OPN compared to controls. Moreover, mutation of the NF-κB binding domain in the HIV-LTR abrogated enhanced promoter activity stimulated by OPN. Interestingly, compared to cerebrospinal fluid from normal and multiple sclerosis controls, OPN levels were significantly higher in HIV-infected individuals both with and without neurocognitive disorder. OPN levels were highest in HIV-infected individuals with moderate to severe cognitive impairment. Moreover, OPN was significantly elevated in brain tissue from HIV-infected individuals with cognitive disorder versus those without impairment. Collectively, these data suggest that OPN stimulates HIV-1 replication and that high levels of OPN are present in the CNS compartment of HIV-infected individuals, reflecting ongoing inflammatory processes at this site despite anti-HIV therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite suppressive anti-HIV therapy, the prevalence of mild HIV-associated neurocognitive disorder (HAND) continues at high levels (Heaton et al. 2010; Simionia et al. 2010). There is a critical need to identify biomarkers and better understand the molecular mechanisms leading to cognitive dysfunction in HAND. Systemic activation of the immune system remains a prominent feature of HIV infection and also affects the inflammatory state of the central nervous system (CNS; Kraft-Terry et al. 2010; Roberts et al. 2010). Activated and/or HIV-infected monocytes trafficking across the blood–brain barrier (Fischer-Smith et al. 2008) and microbial translocation as a result of HIV infection and damage to gut mucosa is thought to be a major source of activating factors (Brenchley et al. 2006) and may play a role in the development of HAND (Fischer-Smith et al. 2008; Ancuta et al. 2008). Moreover, the effectiveness of anti-HIV drug combinations to cross the blood–brain barrier varies significantly and may contribute to viral persistence at this site (Letendre et al. 2008; Best et al. 2009). HIV-1 has evolved an array of strategies to counteract the host immune response. HIV-infected monocytes and viral particles enter the brain where they play a key role in the development of HAND (Giulian et al. 1990; Pulliam et al. 1991; Kaul et al. 2001). While much has been learned about the neurotoxic viral and host factors released from HIV-infected macrophages, a more complete understanding of key cellular pathways that modulate HIV-macrophage interactions and negatively impact neuronal function is needed.
In this regard, we used PCR-mediated subtractive hybridization to enrich for mRNAs upregulated in HIV-infected macrophages. Osteopontin (OPN, protein; SPP1, gene), a cytokine-like phosphoprotein whose over-expression in brain, cerebrospinal fluid (CSF) and plasma, has been associated with neurodegenerative diseases including Parkinson’s (Maetzler et al. 2007; Iczkiewicz et al. 2006), Alzheimer’s (Wung et al. 2007; Comi et al. 2010), multiple sclerosis (Comabella et al. 2005; Vogt et al. 2004), and non-human primate SIV models of HIV infection (Roberts et al. 2003; Burdo et al. 2008) was identified in this screen. We investigated the impact of OPN on HIV replication using knockdown studies in primary macrophages and in a surrogate cell culture model that recapitulates the macrophage phenotype to study the mechanisms by which OPN modulates the viral life cycle. In addition, ex vivo analyses of OPN expression levels in the CSF and brain tissue from HIV-infected individuals with and without cognitive disorder as well as controls was performed. We found that OPN plays a positive role in HIV replication likely through a mechanism leading to an increase in NF-κB activity. Moreover, we found that OPN levels in the CSF of HIV-infected individuals were significantly elevated compared to HIV negative control subjects with non-inflammatory conditions. A significant difference in the level of OPN in the CSF of HIV+ individuals with mild or moderately severe dementia was found. Moreover, in HIV+ individuals with moderately severe dementia, OPN levels were greater than that seen in HIV-uninfected individuals with relapsing remitting multiple sclerosis. Western blot analyses on brain tissue extracts revealed that OPN is significantly elevated in those with moderate to severe cognitive disorder compared to unimpaired subjects. Recent reports show that the receptor for OPN is expressed on specific subsets of neurons (Glezer et al. 2009). Collectively, these data demonstrate that HIV infection leads to an increase in OPN expression that is highest in the CSF and brain of HIV-infected individuals with moderate to severe cognitive impairment suggesting that OPN may play a role in the development of neuronal dysfunction.
Results
OPN message is upregulated in HIV-infected macrophages
Using a PCR-based subtractive hybridization approach we identified the candidate gene SPP1 encoding OPN as an upregulated gene in HIV-infected macrophages. To confirm the differential expression of SPP1, quantitative PCR was performed on RNA isolated from HIV-infected and uninfected macrophages using primers for SPP1 and 18S rRNA as a normalization control for RNA input. OPN mRNA was increased 1.5–1.8-fold in HIV-infected macrophages (one sample t test, p = 0.0001, Fig. 1a). These results confirm the subtractive hybridization results and suggest that SPP1 message is upregulated in HIV-infected macrophages.


The role of osteopontin in HIV replication. a Quantitative PCR for SPP1 and 18S rRNA message was performed on cDNA prepared from HIV-1 SF162, and BaL and infected macrophages and controls. A representative of two independent experiments with mean and standard deviation with three different donors is shown. b Western blot analysis for OPN and actin 48 h after transfection of macrophages with anti-OPN siRNAs (SPP1, 3, 4, 6). c Scatter plots and graph: quantification of GFP+ cells by flow cytometry. A representative of two independent experiments with mean and standard deviation with three different donors is shown. d Western blot analysis for OPN on uninfected TZM-bl cells or cells infected with HIV and recombinant adenovirus expressing OPN or red fluorescent protein (RFP). e Increased replication and syncytia formation in TZM-OPN-expressing cells infected with HIVR3Nef + GFP by fluorescent microscopy and f quantified by flow cytometry (top graph). The mean fluorescent intensity (MFI) of GFP expression in HIV-infected cells or HIV-infected cells over-expressing OPN is shown in the middle graph. Activation of the HIV-LTR-beta-galactosidase promoter is shown in the bottom graph. Means and standard deviations are shown
Knockdown of OPN in primary human macrophages inhibits HIV-1 replication
To determine the role of OPN in HIV replication we used siRNAs directed against OPN to knockdown its expression in primary human MDM. Two of the anti-OPN siRNAs (SPP1 & SPP3) were not effective in diminishing OPN expression and served as internal negative controls for the Western blot analyses (Fig. 1b). Two out of the four siRNAs (SPP4 and SPP6) used nearly completely suppressed OPN expression as assessed by Western blot (Fig. 1b). A scrambled siRNA control (Ctr) was included to rule out inhibition of HIV replication due to any non-specific type 1 interferon response. Under these conditions, HIV replication was decreased nearly threefold (p = 0.0071, Fig. 1c) suggesting that OPN plays a positive role in enhancing virus replication in primary MDM.
OPN enhances HIV-1 replication
Our knockdown studies suggested that OPN might have a stimulatory role in HIV replication. To test this directly, we developed a rapid assay to assess the impact of OPN on HIV replication by expressing OPN in TZM-bl cells using recombinant adenovirus. In contrast to macrophages, TZM-bl cells do not constitutively express OPN (Fig. 1d). Adenoviral particles expressing the red fluorescent protein (RFP) served as a negative control. HIV infectivity as reflected by the number of GFP + cells and syncytium formation was enhanced 2-fold in the presence of recombinant adenovirus expressing OPN (Ad-OPN; p = 0.0084, n = 7; Fig. 1e and f).
In the reporter virus, GFP expression is under the control of the HIV promoter and thus serves as a surrogate marker for viral transcriptional activity (Brown et al. 2005). Analysis of the mean fluorescent intensity of GFP expression revealed significant increases (1.5–2-fold, p = 0.0190, n = 7) in HIV-Ad-OPN- compared to HIV-only infected cells suggesting enhanced transcription of the viral genome (Fig. 1f). TZM-bl cells contain an integrated HIV-LTR promoter fused to the beta-galactosidase gene hence allowing the quantification of promoter activity. In HIV-Ad-OPN-expressing cells compared to HIV-only or HIV-RFP infected cells, a two- to threefold (p = 0.0001) increase in HIV-LTR promoter activity was detected (Fig. 1f). These data suggest that OPN plays a positive role in stimulating HIV replication.
NF-κB activity is increased in HIV-infected cells expressing OPN
The HIV-1 LTR contains two conserved binding sites for NF-κB that function to stimulate HIV transcription (Nabel and Baltimore 1987). The induction of NF-κB in HIV-infected TZM-bl cells expressing OPN could provide a mechanistic explanation for the increases in viral replication observed. To explore this possibility, the activation of NF-κB was measured by examining the degradation of its inhibitor, IκBα at the single-cell level to determine changes in HIV-infected GFP+ cells (Fig. 2a–d; Noursadeghi et al. 2008). Quantitative image analyses showed that in HIV-infected cells levels of IκBα were comparable to uninfected cells. An 11% reduction of IκBα was seen in cells expressing OPN suggesting that OPN alone can stimulate signaling pathways involved in IκBα degradation (Fig. 2, graph). In HIV-Ad-OPN infected cells a further reduction in IκBα expression of 20% (p = 0.0001) compared to uninfected cells was seen suggesting an additive effect of OPN and HIV infection in the activation of IκBα degradation (Fig. 2, graph). A threefold (p = 0.001) decrease in IκBα expression was also detected by Western blot analyses on HIVSF162-infected, OPN-expressing cells compared to cells infected with Ad-RFP (Fig. 2). Expression of OPN in HIV-infected cells led to an 1.4-fold increase in the nuclear translocation of NF-κB as reflected in the nuclear:cytoplasmic ratio of NF-κB in HIV-Ad-OPN infected cells compared to uninfected cells (p = 0.0001; Fig. 3a, graph). To obtain more direct evidence for the putative modulation of HIV transcription by OPN, HIV-LTR chloramphenicol acetylase (CAT) reporter constructs having an intact or mutated NF-κB site were co-transfected into TZM-bl cells with control or a plasmid-encoding OPN. A significant activation of CAT enzyme activity in the presence of OPN expression was detected only when the LTR contained an intact NF-κB site (Fig. 3b, p = 0.0001). Together, these results suggest that expression of OPN in HIV target cells stimulates a signal transduction pathway leading to the upregulation of NF-κB activity and, importantly, results in the enhancement of HIV replication.

Single-cell analyses reveal that the NF-κB inhibitor IκBα is decreased in HIV-infected cells expressing OPN. TZM-bl cells infected with HIVR3Nef + GFP (green) were fixed and immunostained for IκBα (red), nuclei with DRAQ5 (blue). a TZM-bl cells only; b TZM-bl infected with Ad-OPN; (c) HIV-infected TZM-bl; d HIV-infected TZM-bl expressing OPN. The relative normalized pixel intensity of IκBα is shown (top right graph). All pictures were taken on a LSM 510 Meta at the same pinhole and intensity settings. The means and standard deviations of a representative of two independent experiments are shown. Western blot analysis for IκBα is shown in the middle panel and quantification shown in the bottom graph: HIV-infected TZM-bl cells only (HIV) or co-infected with Ad-RFP (HIVRFP) or Ad-OPN (HIVOPN); HIV-uninfected cells and exposed to Ad-RFP (RFP) or Ad-OPN (OPN). A representative of two independent experiments is shown

NF-κB activity is increased in HIV-infected cells expressing OPN. TZM-bl cells infected with HIVR3Nef + GFP were fixed and immunostained for NFκB (red) and nuclei with DRAQ5 (blue). a TZM-bl cells only; b HIV-infected TZM-bl; c HIV-infected TZM-bl expressing OPN; d TZM-bl infected with Ad-OPN; (I) the relative normalized nuclear:cytoplasmic intensity of NFκB is shown. All pictures were taken on a LSM510 Meta at the same pinhole and intensity settings. (II) TZM-bl cells were co-transfected with HIV-LTR-CAT reporter constructs encoding an intact (pHIV-CAT) or mutated NFκB (pDkB-HIV-CAT) binding domain and control (pRC) or OPN-expressing plasmid (OPN). CAT protein was measured by ELISA. The means and standard deviations of a representative of two independent experiments are shown
OPN levels are significantly increased in the CSF of HIV-infected persons and exacerbated in those with severe cognitive disorder
While our in vitro data suggested that OPN is increased in HIV-infected macrophages, we wanted to determine whether such a change could be detected in plasma and CSF from HIV-infected individuals with and without cognitive disorder. We used stored CSF from the Northeast AIDS Dementia (NEAD) Cohort, a group containing individuals with advanced disease (Marder et al. 2003; Sacktor et al. 2002; Table 1). The NEAD Cohort was a longitudinal multicenter cohort study in which individuals underwent serial examination with neurocognitive testing and body fluid sampling as reported previously (Marder et al. 2003; Sacktor et al. 2002). The degree of cognitive impairment was assessed using the MSK (Price and Brew 1988). CSF samples from HIV-uninfected individuals with either non-inflammatory or inflammatory diagnoses were used as controls (Table 1). No significant differences in OPN plasma levels of HIV-infected individuals with and without cognitive disorder were found, but differences in mean OPN concentration in the CSF were detected (Fig. 4a). Mean OPN levels were significantly higher in HIV-infected persons without cognitive disorder (MSK 0) than in the CSF from uninfected persons with non-inflammatory conditions (Table 1; p = 0.05, Fig. 4a). In persons with moderately severe HIV-associated cognitive disorder (MSK 2), mean OPN CSF levels were significantly higher than those with no or less severe impairment (p = 0.01 to 0.05; Fig. 4a). Moreover, mean OPN CSF levels in the MSK 2 group were significantly increased compared to CSF levels from HIV-uninfected patients diagnosed with inflammatory diseases including relapsing remitting and secondary progressive multiple sclerosis (p = 0.001; Fig. 4; Table 1). No biologically significant associations were found between OPN levels and plasma or CSF HIV viral load, CD4 T-cell count or age (Fig. 5).

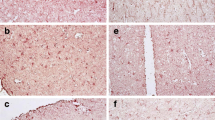
OPN is increased in the CSF and brain of HIV-infected persons. a Samples were grouped by MSK with MSK 0 (n = 21), representing normal cognition and MSK 0.5 (n = 35), MSK 1 (n = 33) and MSK 2 (n = 13) representing increasing severity of cognitive impairment; population means are indicated by hash mark. Inflammatory (n = 30) and non-inflammatory (n = 27) CSF samples were obtained from HIV-uninfected individuals (Table 1). Data was analyzed with one-way ANOVA, Dunn’s multiple comparison post test: *p < 0.05, **p < 0.01, ***p < 0.001. b Osteopontin is increased in brain tissue of HIV-infected persons with cognitive disorder. Cell lysates prepared from frozen brain tissue from HIV-infected patients with stable neuropsychological diagnoses from the NNTC (Table 2, supplemental data) were analyzed by Western blot for OPN and β3-tubulin and quantified (c). HAD HIV-associated dementia, MCMD minor cognitive motor disorder, Sub subsyndromic, Normal neurocognitively normal, Cont control TZM-bl lysate. The Westerns were also probed for the HIV Nef protein. A representative of three independent experiments is shown

No biologically significant associations between CSF OPN levels and plasma or CSF HIV viral load, CD4 T-cell count or age. Correlation analyses were determined assuming sampling from a Gaussian distribution. Pearson r and R 2 are indicated
OPN is increased in brain tissue of HIV-infected persons with cognitive disorder
Frozen brain tissue samples from the occipital lobes were obtained from the National NeuroAIDS Tissue Consortium (NNTC, Table 2, supporting data; Morgello et al. 2001; Everall et al. 2009). The NNTC uses the Psychiatric Research Interview for Substance and Mental Disorders to define cognitive disorder. The NNTC’s primary diagnostic diagnosis equates to the MSK rating scales as follows: MSK 0 = neurocognitively normal, MSK 0.5 = subsyndromic, MSK 1 = possible or probable minor cognitive motor disorder (MCMD) and MSK 2 to 4 = possible or probable HIV-associated dementia ranked by neurological severity. Western blot analyses revealed a marked and significant increase in OPN levels of persons with more severe HAND compared to HIV-infected individuals without or with mild cognitive impairment (p = 0.0387; Fig. 4b, c). The HIV Nef protein plays an important role in viral pathogenesis. However, we found that the increase in OPN detected in brain tissues was not related to the presence or absence of HIV Nef expression as no significant differences in Nef levels were found between groups (Fig. 4b).
Discussion
Osteopontin is a 314-amino-acid protein first reported as a secreted phosphoprotein (SPP1) upregulated in cells after neoplastic transformation (Craig et al. 1988). It was also described as the cytokine, early T-lymphocyte activation marker-1 (Eta-1) involved in the induction of Th1-cell-mediated immunity (Patarca et al. 1989). OPN is modified post-translationally generating proteins in the range of 44–75 kDa on denaturing SDS-PAGE (Denhardt and Guo 1993). A thrombin cleavage site, when used, generates two halves of OPN that retain distinct biological functions (Ashkar et al. 2000). Interestingly, while OPN function confers cellular immunity to intracellular pathogens such as mycobacterial species, Rickettsia tsutsugamushi, Listeria monocytogenes, Flavivirus, Rotavirus, and herpes simplex virus type 1, its over-expression in autoimmune disorders and hepatitis infection exacerbates disease (Lampe et al. 1991; Sato et al. 2005; Wong et al. 2005; Saito et al. 2007). Interestingly, OPN was shown to promote the survival of myelin-reactive T-cells by inhibiting the transcription factor Foxo3a, activating NF-κB, and blocking genes regulating apoptosis (Hur et al. 2006). Our study is the first to show that OPN has a direct effect on HIV-1 replication. We found that knockdown of OPN in macrophages significantly impairs HIV replication suggesting, in contrast to several other intracellular pathogens, a positive role for OPN. Indeed, our data show that OPN significantly enhances HIV replication and suggests that HIV has hijacked an OPN-mediated signaling pathway leading to the activation of NF-κB to enhance viral replication and spread. Based on our findings, we would expect that OPN is acting on cellular pathways that activate cells and in turn create a microenvironment more efficient for reverse transcription and viral transcription. A better understanding of the molecular mechanisms governing OPN-HIV interaction may allow the development of strategies to interfere with virus replication in macrophages, a cell type that serves as a persistent viral reservoir.
Inflammation is a prominent component of Parkinson’s, Alzheimer’s, and multiple sclerosis CNS diseases that may either proceed or result from the progression of neurodegenerative processes. HIV CNS disease is also characterized by an enhanced inflammatory response with increased MCP-1, IL-1β, IL-6, IFN-γ, IL-15, and glia cell activation (Kraft-Terry et al. 2010). Evidence demonstrating that lentiviral infection of the brain induces an increase in OPN was recently reported for non-human primate models of SIV/HIV infection (Roberts et al. 2003; Burdo et al. 2008). One study reported that increased OPN levels in the plasma of HIV-infected individuals correlated with cognitive impairment compared to those without HAND, however, OPN levels in the CSF did not differ between the groups (Burdo et al. 2008). In contrast to this study, we saw no significant differences in levels of plasma OPN between neurocognitive groups, but did observe increased levels in the CSF, particularly in those HIV-infected persons with mild (MSK 1) and moderate cognitive disorder (MSK 2). One potential explanation for the disparate results is that the OPN transcript is subject to alternative splicing and can also be cleaved into biological active N-terminal and C-terminal halves. Commercially available ELISA kits used in the Burdo et al. study are not designed to differentiate between OPN variants. Indeed, we found significant variations in the ability of commercially available mouse monoclonal antibodies to serve as efficient capture agents in our ELISA. Extensive variation in quantifying OPN levels using has been reported (Anborgh et al. 2009).
Our finding that HIV infection of macrophages upregulates OPN would suggest a potential mechanism by which OPN is increased in vivo. In several pathological conditions in which activated T-lymphocytes and macrophages play a role such as rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, and atherosclerosis, all are associated with high tissue and plasma OPN. Monocytes, particularly the CD14+CD16+ subset, migrating from the bone marrow to blood to the CNS, are believed to play a key role in seeding the brain with HIV as well as contributing to inflammation at this site (Koenig et al. 1986; Fischer-Smith et al. 2001; Pulliam et al. 1997; Crowe et al. 2003). In this regard, OPN is a known chemoattractant for monocytes and additionally can prevent monocyte recirculation and apoptosis (Burdo et al. 2007). Interestingly, Marcondes et al., found that increased expression of the OPN receptor CD44v6 on both CD14+CD16low and CD14+CD16hi monocytes could distinguish among groups of animals with or without SIV encephalitis (Marcondes et al. 2008). These findings suggest a possible role of OPN in the recruitment of activated monocytes to the brain. In this regard, we found that OPN is significantly increased in brain tissue of HIV-infected patients with cognitive disorder compared to those with normal cognition. The cells responsible for increased OPN expression in the brain would include macrophages and microglia; however, neurons can also produce the protein (Yasuto et al. 2009). Moreover, specific subsets of neurons were recently shown to express the OPN receptor, CD44 suggesting the real possibility that OPN can directly modulate neuronal function (Glezer et al. 2009).
Collectively, our data lend support for the idea that OPN levels in the CSF and brain are exacerbated in those with moderate HIV-associated dementia and might reflect the accumulation of persistent inflammatory insults that lead to neurodegeneration and neuronal dysfunction. Indeed, our assay results suggest that similar levels of OPN would be expected in side-by-side comparisons of CSF of patients with HAND or multiple sclerosis. Although the incidence of HIV-infected persons with severe cognitive disorder has greatly decreased, early changes in the level of OPN in the CNS may be indicative of the progression of inflammation at this site and could potentially be used in combination with other markers as an early indicator for probable CNS dysfunction. Moreover, as HIV-1 infection can increase OPN, the detection of elevated levels of this cytokine in the CNS may be indicative of high levels of ongoing viral replication at this site. In the HIV-R3Nef + GFP reporter virus, the GFP gene is located in the Nef position followed by an IRES element driving Nef expression. We have shown in long-term cultures (60–78 days) of primary macrophages infected with this virus that a subpopulation of cells remain transcriptionally active and continue to express GFP and Nef although they make no capsid protein (Brown et al. 2006). Indeed, the early gene HIV Nef was readily detected in brain tissue. Future studies will focus on testing and exploring these ideas.
Materials and methods
Macrophage culture and subtractive hybridization
Primary human monocyte-derived macrophages were infected with HIVR3Nef + GFP and harvested as previously described (Brown et al. 2006). Total RNA was isolated from sorted cells and amplified by long-distance PCR as directed by the manufacturer (PCR-Select cDNA Subtraction Kit, Clontech). Subtracted cDNAs were size- fractionated and subcloned into the vector pCR2.1 and sequenced.
Quantitative PCR
Quantitative PCR for SPP1 (accession number BCO17387) and 18S rRNA (accession number NT_167214.1) was performed using 2.5 pmol of each of the following primers SPP1-F: 5′AGCGAGGAGTTGAATGGTG, SPP1-R: 5′GCTTGTGGCTGTGGGTT; 18S rRNA-F: 5′CTCAACACGGGAAACCTCAC and 18S rRNA-R: 5′AGACAAATCGCTCCACCAAC in a 25 μl reaction with Power SYBR green and analyzed on the ABI Prism 7000 Sequence Detection System (Applied Biosystems).
Knockdown of OPN in primary human macrophages
Macrophages differentiated for 5 days were detached, washed twice with PBS, and resuspended at 2 × 105 cells/well in 10 μl/well of resuspension buffer and 50–80 pmol of anti-OPN or the Allstars scrambled negative control siRNAs in triplicates (Qiagen) and transfected using the Neon Transfection System (Invitrogen). Transfection of rhodamine-labeled negative control siRNA was used to monitor the efficiency of RNA uptake. The macrophages were infected with HIVR3Nef + GFP overnight at 48 h post-transfection and harvested 3 days later and analyzed by flow cytometry.
HIV replication assays
TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program from J. Kappes and X. Wu and Tranzyme Inc. and cultured in DMEM medium containing 10% FBS. TZM-bl cells (105) were plated on fibronectin-coated 24-well plates and the next day infected in triplicate for 3 h with 106 adenoviral particles per well. The cells were then infected with HIVR3Nef + GFP in DMEM/2%FBS/4 μg/ml DEAE-dextran overnight. The next day, the inoculum was removed and replaced with 1 ml of DMEM/10%FBS. After 48–72 h post-infection, cells were harvested for flow cytometry.
Cloning of recombinant adenovirus
The full-length SPP1 cDNA (OPN; Origene, TC127388) was cloned into pAd/CMV/V5-DEST to generate replication defective adenoviral particles expressing SPP1 (Invitrogen). Adenoviral particles were purified using the VivaPack AdenoPure kit (Sartorius), titered with the Adeno-X Rapid Titer Kit (Clontech) and stored in aliquots at −80°C. Adenoviral-RFP particles expressing DsRed2 were obtained from Vector Biolabs.
Confocal microscopy and quantitative image analysis
TZM-bl cells were immunostained with anti-NF-κB antibody (#436700, Invitrogen), or IκBα (cat# 4814, Cell Signaling), and incubated for 3–5 min with a 1:50 dilution of DRAQ5 (Biostatus Limited). The normalized intensity values for IκBα staining were calculated for three fields of confluent cells using Image J software. The nuclear:cytoplasmic ratio of NF-κB signal was calculated as described (Noursadeghi et al. 2008). A median filter was applied followed by the creation of threshold masks for the nuclear and cytoplasmic regions of interest. The normalized intensity values for the nuclei and cytoplasm were calculated and used to generate the nuclear:cytoplasmic ratios and to allow comparisons between experiments.
CAT ELISA
TZM-bl were transfected using lipofectamine 2000 (Invitrogen) according to the manufacturers suggestion with pHIV-CAT or pDkB-HIV-CAT (obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NAID, NIH: from Dr. Gary Nabel and Neil Perkins) and pRC-CMV or a plasmid-encoding osteopontin (pCMV-OPN, TrueClone, Origene). CAT enzyme protein was quantified using an ELISA method (Roche). Protein concentration was determined by Bradford assay (Biorad) and used to normalize the CAT assay results.
OPN ELISA
Plasma and CSF samples were obtained from the Johns Hopkins Neurology Plasma and CSF Tissue Bank and stored in aliquots at −80°C. Monoclonal antibody to OPN (MAB194, Maine Biotechnology) was used at 5 μg/ml to coat 96-well plates (Immubulon). Rabbit anti-OPN (FL-314, Santa Cruz) served as the detection antibody at 1:1,000. Recombinant OPN (R&D Systems) was used to generate the standard curves. All antibody incubations were performed in PBS-1%Tween containing a protease inhibitor cocktail. Antibody binding was detected with goat-anti-rabbit-HRP and developed with TMB substrate solution. Samples were assayed in duplicate and repeated two to three times. Data was analyzed with one-way ANOVA, and Dunn’s multiple comparison post test using Graphpad PRISM. Correlation analyses were determined assuming that the values were sampled from populations that follow a Gaussian distribution by Pearson r and R 2.
Western analyses
Brain tissue was obtained from the National NeuroAIDS Tissue Consortium (NNTC; Morgello et al. 2001; Everall et al. 2009) and handled under biohazard level 2 safety conditions as approved by the Johns Hopkins Institutions Biosafety Office. Frozen tissue was solubilized in 50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 5 mM EDTA, 1% sodium deoxycholate, 10% glycerol, 1 mM DTT, protease inhibitor cocktail and 10 mM sodium vanadate. Aliquots were stored at −80°C. NuPAGE 4–12% SDS Bis–Tris gels were run in MOPS buffer and blotted to nitrocellulose membranes using the iBlot System (Invitrogen). ECL Plus reagents (GE Healthcare) were used to develop the blots.
References
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17:3–16
Simionia S, Cavassinic M, Annonia J-M, Abrahame AR, Bourquina I, Schifferf V, Calmyf A, Chaved J-P, Giacobinig E, Hirschelf B, Du Pasquiera RA (2010) Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 24:1243–1250
Kraft-Terry SD, Stothert AR, Buch SJ, Gendelman HE (2010) HIV-1 neuroimmunity in the era of antiretroviral therapy. Neurobiol Dis 37:524–548
Roberts TK, Buckner CM, Berman JW (2010) Leukocyte transmigration across the blood–brain barrier: perspectives on neuroAIDS. Front Biosci 15:478–536
Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J (2008) Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol 14:318–326
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371
Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D (2008) Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE 3:e2516
Letendre S, Marquie-Beck J, Capparelli E, Best B, Clifford D, Collier AC, Gelman BB, McArthur JC, McCutchan JA, Morgello S, Simpson D, Grant I, Ellis RJ, Group, C (2008) Validation of the CNS penetration-effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol 65:65–70
Best BM, Letendre SL, Brigid E, Clifford DB, Collier AC, Gelman BB, McArthur JC, McCutchan JA, Simpson DM, Ellis R, Capparelli EV, Grant I, Group, C (2009) Low atazanavir concentrations in cerebrospinal fluid. AIDS 23:83–87
Giulian D, Vaca K, Noonan CA (1990) Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 250:1593–1596
Pulliam L, Herndler BG, Tanf NM, McGrath MS (1991) Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. J Clin Invest 87:503–512
Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994
Maetzler W, Berg D, Schalamberidze N, Melms A, Schott K, Mueller JC (2007) Osteopontin is elevated in Parkinson's disease and its absence leads to reduced neurodegeneration in the MPTP model. Neurobiol Dis 25:473–482
Iczkiewicz J, Jackson MJ, Smith LA, Rose S, Jenner P (2006) Osteopontin expression in substantia nigra in MPTP-treated primates and in Parkinson's disease. Brain Res 1118:239–250
Wung JK, Perry G, Kowalski A, Harris PL, Bishop GM, Trivedi MA, Johnson SC, Smith MA, Denhardt DT, Atwood CS (2007) Increased expression of the remodeling- and tumorigenic-associated factor osteopontin in pyramidal neurons of the Alzheimer's disease brain. Curr Alzheimer Res 4:67–72
Comi C, Carecchio M, Chiocchetti A, Nicola S, Galimberti D, Fenoglio C, Cappellano G, Monaco F, Scarpini E, Dianzani U (2010) Osteopontin is increased in the cerebrospinal fluid of patients with Alzheimer's disease and its levels correlate with cognitive decline. J Alzheimers Dis 19:1143–1148
Comabella M, Pericot I, Goertsches R, Nos C, Castillo M, Blas Navarro J, Rio J, Montalban X (2005) Plasma osteopontin levels in multiple sclerosis. J Neuroimmunol 158:231–239
Vogt MH, Floris S, Killestein J, Knol DL, Smits M, Barkhof F, Polman CH, Nagelkerken L (2004) Osteopontin levels and increased disease activity in relapsing remitting multiple sclerosis patients. J Neuroimmunol 155:155–160
Roberts ES, Zandonatti MA, Watry DD, Madden LJ, Henriksen SJ, Taffe MA, Fox HS (2003) Induction of pathogenic sets of genes in macrophages and neurons in NeuroAIDS. Am J Pathol 162:2041–2057
Burdo TH, Ellis RJ, Fox HS (2008) Osteopontin is increased in HIV-associated dementia. J Infect Dis 198:715–722
Glezer I, Bittencourt JC, Rivest S (2009) Neuronal expression of Cd36, Cd44, and Cd83 antigen transcripts maps to distinct and specific murine brain circuits. J Comp Neurol 517:906–924
Brown A, Gartner S, Kawano T, Benoit N, Cheng-Mayer C (2005) HLA-A2 down-regulation on primary human macrophages infected with an M-tropic EGFP-tagged HIV-1 reporter virus. J Leukoc Biol 78:675–685
Nabel G, Baltimore D (1987) An inducible transcription factor activates expression of human immunodeficiency virus in T-cells. Nature 326:711–713
Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR (2008) Quantitative imaging assay for NF-κB nuclear translocation in primary human macrophages. J Immunol Methods 329:194–200
Marder K, Albert SM, McDermott MP, McArthur JC, Schifitto G, Selnes OA, Sacktor N, Stern Y, Palumbo D, Kieburtz K, Cohen B, Orme C, Epstein LG (2003) Inter-rater reliability of a clinical staging of HIV-associated cognitive impairment. Neurology 60:1467–1473
Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L (2002) HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol 8:136–142
Price RW, Brew BJ (1988) The AIDS dementia complex. J Infect Dis 158:1079–1083
Morgello S, Gelman BB, Kozlowski PB, Vinters HV, Masliah E, Cornford M, Cavert W, Marra CM, Grant I, Singer EJ (2001) The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol 27:326–335
Everall I, Vaida F, Khanlou N, Lazzaretto D, Achim C, Letendre SL, Moore D, Ellis RJ, Cherner M, Gelman BB, Morgello S, Singer EJ, Grant I, Masliah E, (NNTC), NNTC, (2009) Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol 15:360–370
Craig AM, Nemir M, Mukherjee BB, Chambers AF, Denhardt DT (1988) Identification of the major phosphoprotein secreted by many rodent cell lines as 2ar/osteopontin: enhancec expression in H-ras-transformed 3 T3 cells. Biochem Biophys Res Commun 157:166–173
Patarca R, Freeman GJ, Singh RP, Wei F-Y, Durfee T, Blattner F, Regnier DC, Kozak CA, Mock BA, Morse HC, Jerrells TR, Cantor H (1989) Structural and functional studies of the early T-lymphocyte activation 1 (Eta-1) gene. J Exp Med 170:145–161
Denhardt DT, Guo X (1993) Osteopontin: a protein with diverse functions. FASEB J 7:1475–1482
Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H (2000) Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287:860–864
Lampe MA, Patarca R, Iregui MV, Cantor H (1991) Polyclonal B cell ativation by the Eta-1 cytokine and the development of systemic autoimmune disease. J Immunol 147:2902–2906
Sato T, Nakai T, Tamura N, Okamoto S, Matsuoka K, Sakuraba A, Fukushima T, Uede T, Hibi T (2005) Osteopontin/Eta-1 upregulated in Crohn's disease regulates the Th1 immune response. Gut 54:1254–1262
Wong CK, Lit LC, Tam LS, Li EK, Lam CW (2005) Elevation of plasma osteopontin concentration is correlated with disease activity in patients with systemic lupus erythematosus. Rheumatology 44:602–606
Saito Y, Kon S, Fujiwara Y, Nakayama Y, Kurotaki D, Fukuda N, Kimura C, Kanayama M, Ito K, Diao H, Matsui Y, Komatsu Y, Ohtsuka E, Uede T (2007) Osteopontin small interfering RNA protects mice from fulminant hepatitis. Hum Gene Ther 18:1205–1214
Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L (2006) Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol 8:74–83
Anborgh PH, Wilson SM, Tuck AB, Winquist E, Schmidt N, Hart R, Kon S, Maeda M, Uede T, Stitt LW, Chambers AF (2009) New dual monoclonal ELISA for measuring plasma osteopontin as a biomarker associated with survival in prostate cancer: clinical validation and comparison of multiple ELISAs. Clin Chem 55:895–903
Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS (1986) Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 233:1089–1093
Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L'Heureux D, Régulier EG, Richardson MW, Amini S, Morgello S, Khalili K, Rappaport J (2001) CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol 7:528–541
Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS (1997) Unique monocyte subset in patients with AIDS dementia. Lancet 349:692–695
Crowe SM, Zhu T, Muller WA (2003) The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J Leukoc Biol 74:635–641
Burdo TH, Wood MR, Fox HS (2007) Osteopontin prevents monocyte recirculation and apoptosis. J Leukoc Biol 81:1504–1511
Marcondes MCG, Lanigan CMS, Burdo TH, Watry DD, Fox HS (2008) Increased expression of monocyte CD44v6 correlates with the development of encephalitis in rhesus macaques infected with simian immunodeficiency virus. J Infect Dis 197:1567–1576
Yasuto K, Shin-ichi N, Yoshiaki H, Masahiro M, Tsutomu S, Toshimitsu S (2009) The immunohistochemical expression profi le of osteopontin in normal human tissues using two site-specifi c antibodies reveals a wide distribution of positive cells and extensive expression in the central and peripheral nervous systems. Med Mol Morphol 42:155–161
Brown A, Zhang H, Lopez P, Pardo CP, Gartner S (2006) In vitro modeling of the HIV-macrophage reservoir. J Leukoc Biol 80:1127–1135
Acknowledgments
We thank the Margaret Q. Landenberger Foundation, the Campbell Foundation, and the Cooke Family Foundation for funding this work. We also thank Hao Zhang from the Flow Cytometry Core at the Johns Hopkins School of Public Health and Amy Kosel, Liping Guo, Peter Dziedzic, and Jason Creighton for technical assistance. The NEAD cohort was supported through NS44807 (JCM) and the data analysis and statistical review was supported through 1P30MH075673 (JCM). We thank The National NeuroAIDS Tissue Consortium supported through N01MH32002 for providing the brain tissue samples.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table 2
Clinical Characteristics of Brain Tissue Samples (DOC 47 kb)
Rights and permissions
About this article
Cite this article
Brown, A., Islam, T., Adams, R. et al. Osteopontin enhances HIV replication and is increased in the brain and cerebrospinal fluid of HIV-infected individuals. J. Neurovirol. 17, 382–392 (2011). https://doi.org/10.1007/s13365-011-0035-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-011-0035-4