
Abstract
Non-invasive genetic sampling using materials such as faeces or hair can be used to monitor wildlife populations, although DNA quality is often poor. Improving sampling efficiency and minimising factors that reduce DNA quality are therefore critical. After a severe decline, the European pine marten, Martes martes, has reclaimed much of its former range in Scotland, UK. Recording this rapid range expansion requires developing techniques for accurate monitoring, but this is hampered by the species’ elusive behaviour. We tested two sampling methods, hair collected from hair tubes and faeces (scat) collected along tracks, to assess the effects of key environmental and sampling variables on DNA quality and sampling efficiency. For hair, we tested the influence of hair tube location (distance from forest tracks) on collection rate and sex ratio of animals successfully sampled. For scats, we assessed the effect of time since defecation (1 to 16 days) on genotyping error rates and success under two contrasting environmental conditions (exposed to rainfall or sheltered). We found no bias in the collection rate or sex ratio of animals detected by hair samples with differing proximity to forest tracks. DNA amplification failure for scats exposed to rainfall increased from 28 to 65 % over the 16-day experimental period. During periods of low rainfall, the length of collection sessions could therefore be extended to increase sample number without risk of DNA degradation. Lack of bias in hair collection rates with proximity to forest tracks provides justification for tube placement close to tracks, as this reduces survey effort. These findings provide guidance for the development of efficient and cost-effective non-invasive sampling of Scottish pine martens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Accurate baseline data on species presence, abundance and demographic rates is a key component of effective wildlife management (Gibbs et al. 1999). For rare or threatened species, knowledge of population status enables informed management decisions to be made and adaptive conservation relies on the ability to monitor the effects of management (Head et al. 2013). In order to monitor species of conservation concern, there must be a reliable method of detection. Traditional methods of detection often involve capturing animals, which can be difficult when species are elusive or protected and stressful for animals vulnerable to disturbance. Non-invasive genetic sampling has been suggested as an alternative survey tool, with genetic samples extracted from hair, faeces or feathers potentially negating the need to physically capture or even observe the animal (Taberlet et al. 1996; Taberlet and Luikart 1999). To date, non-invasive DNA methods have been used for a range of purposes including mapping distributions (e.g. the Andean cat in Peru, Orealilurus jacobita, Cossios et al. 2007; jaguar in Belize, Panthera onca, Weckel et al. 2006), estimation of population densities (e.g. coyote, Canis latrans, Kohn et al. 1999; the ship rat, Rattus rattus, Wilson et al. 2007) and comparisons of survival estimates between the sexes (e.g. Wolverine populations Gulo gulo, Brøseth et al. 2010).
Genetic methods, however, are not without drawbacks. Sample processing is costly and, in the case of wide-ranging or low density populations, collecting sufficient samples can also be time consuming and expensive. These issues may be exacerbated when using samples of poor quality DNA such as faeces (Lucchini et al. 2002), which contain compounds that inhibit the DNA amplification process. DNA quality is measured by the rate at which amplification, through polymerase chain reaction (PCR), yields a detectable quantity of DNA, quantified as PCR ‘success’ or ‘failure’ rate; and the rate of occurrence of amplification errors. Two types of error are prominent: allelic dropout, where one allele from a heterozygous individual fails to amplify; and false alleles, where an allele differing from the consensus, or agreed, genotype is produced (Broquet et al. 2007). For practices which only require identification at species level, such as distribution mapping, researchers may be concerned with maximising the rate of PCR success but, once a sample has been genotyped with a species specific marker, the occurrence of error within this marker will be largely unimportant. For studies requiring individual identification, such as estimates of population density, error rates must also be considered and minimised. In these cases, data with an acceptable level of precision may only be achieved through larger sample sizes and repeated amplifications, as well as through the use of more expensive DNA extraction techniques (Taberlet et al. 1996). Improving the efficiency of sampling and minimising the factors that reduce DNA quality are therefore critical when designing a cost-effective surveying strategy.
Despite previous findings that suggest a decrease in faecal DNA quality over time (Brinkman et al. 2010; Panasci et al. 2011), and with increased rainfall (Nsubuga et al. 2004; Murphy et al. 2007; Brinkman et al. 2010), there is considerable variation in the effect of these factors between taxa. For example, rainfall significantly degrades DNA in Sitka black-tailed deer pellets (Odocoileus hemionus sitkensis), but does not affect DNA sample quality from mountain gorilla faeces (Beringei beringei; Nsubuga et al. 2004). Similarly, amplification success as faecal samples aged (up to 1 month) decreased by 65 % for the brush-tailed rock-wallaby (Petrogale penicillata; Piggott and Taylor 2003), but only 5 % for coyote (Canis latrans; Panasci et al. 2011). Genotyping success has been higher for hair samples than scats for pine martens in previous studies (Mullins et al. 2009), but success rates for hair can still vary, with factors such as the number of hairs that are used in the extraction process having a significant effect, as seen for the Asiatic black bear (Ursus tibetanus, Uno et al. 2012), although it remains unclear if differences exist between species.
Pine marten populations in Scotland have shown a recent range expansion after near-extinction in the early twentieth century (Lockie 1964; Croose et al. 2013). As a protected native species, there is strong stakeholder interest in the conservation of pine martens, particularly since the suggestion that they may play a role in controlling the invasive American grey squirrel (Sciurus carolinensis; Sheehy and Lawton 2014). There is concern, however, about the effect of pine martens on vulnerable prey species through, for example, nest predation of capercaillie (Tetrao urogallus) populations (Summers et al. 2009). Their elusive behaviour makes non-invasive sampling such as DNA extraction from hair or faeces potentially useful. Genetic analyses of scat have been successfully used for species identification and for determining the distribution of martens in Scotland (Caryl et al. 2012a; Croose et al. 2013) but have thus far been unsuccessful in individual-level analyses due to poor quality DNA. This has prompted the need for an assessment of the factors affecting DNA quality in order for these factors to be minimised in future studies.
Sampling regimes used to estimate population abundance and density should account for differences in detectability, either through sampling design or through statistical methods. For studies using non-invasive hair sampling, time constraints usually make it unfeasible to relocate hair tubes between sampling sessions, which may introduce a temporal bias and violate assumptions of sampling independence (Boulanger et al. 2006). For example, heterogeneity in the probability of capture between individual pine marten has been observed in an Irish study, with hair tubes placed in lowland forests collecting more samples than those in upland forests, despite similar population densities in both habitats (Lynch et al. 2006). Spatial biases can also occur; hair tubes are most accessible if placed close to forest tracks; pine marten scats are also collected from forest tracks due to the relative ease of collection compared to searching the densely vegetated, forest floor. If some individuals use forest tracks less frequently than others, the samples collected may only represent a sub-set of the population. Female pine martens, for instance, are thought to be more risk averse than males due to the reporting of a higher proportion of male road casualties (Rob Coope, personal communication); females also maintain smaller home ranges than males (Caryl et al. 2012b), which therefore could be less likely to contain forest tracks. As a consequence, the effect of different sampling techniques and designs on the outcome of non-invasive hair sampling is currently unclear.
In this paper we assess the effects of key environmental and sampling variables on the quality of pine marten DNA sampled non-invasively through hair and scats (with the latter divided into experimental treatments to test for the effect of exposure to rainfall) and examine the implications for developing efficient sampling protocols. Specifically, we address the following questions:
-
1.
How does time (measured as consecutive sampling sessions) influence hair tube sample independence (hair samples only)?
-
2.
Does distance from forest track affect the visitation rates of pine marten, and does this vary between the sexes (hair samples only)?
-
3.
How is PCR success affected by the number of hair follicles included in the reaction (hair samples only)?
-
4.
What are the effects of time since defecation and exposure to rainfall on DNA genotyping success and error rates (scats only)?
Materials and methods
Study areas
Four forests in the Scottish Highlands known to have pine martens present were surveyed. Abernethy Forest National Nature Reserve (57°15′N, 3°40′W; hereafter Abernethy) is a Royal Society for the Protection of Birds (RSPB) reserve in the northern Cairngorms covering 36 km2 of both ancient native pinewood (approx. 24 km2) and Scots pine (Pinus sylvestris) plantation (Summers et al. 2010). Mar Lodge Estate (57°00′N, 3°37′W; hereafter Mar), owned by the National Trust for Scotland, comprises Caledonian pinewood concentrated mainly along Glen Lui and Glen Quioch, north west of Braemar (Davies and Legg 2008). Inshriach Forest (57°06′N, 3°56′W, hereafter Inshriach) is a Forestry Commission owned site in the Northern Cairngorms consisting mainly of managed Scots pine plantation with some remnants of Caledonian pinewood (Twiddle and Quine 2011). Darnaway Forest (57°33′N, 3°45′W; hereafter Darnaway), which is managed by Moray Development Company Ltd, consists of commercial Scots pine, Sitka spruce (Picea sitchensis) and Douglas fir (Pseudotsuga sp.) plantation, with some areas of deciduous woodland.
Sample collection
Hair was sampled during September to November at two forests in 2011 (Abernethy, Mar) and two forests in 2012 (Darnaway, Inshriach) using hair tubes fitted with sticky pads (Mullins et al. 2009) and labelled with a unique identifier (Hairtube ID). Four sampling sessions were held in Abernethy and Darnaway, and five each at Inshriach and Mar (Online resource 1), with each session taking five (Mar, Inshriach) or six consecutive days (Darnaway, Abernethy). Hair samples from each tube were collected in individual polythene bags and labelled with a unique identifier. All samples were frozen at −20 °C within 8 h and transferred to −80 °C within 3 weeks to await DNA analysis.
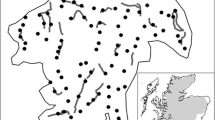
Hair tube placement within each forest was planned using 1:25,000 Ordnance Survey maps. To ensure that at least one hair tube was placed in each potential home range (Caryl et al. 2012b), one (Abernethy, Mar) or two (Inshriach, Darnaway) hair tubes were placed in each 1 km2 grid cell within the study area (Fig. 1), giving a total of 33 hair tubes at Abernethy, 26 at Mar, 64 at Inshriach and 47 in Darnaway. For ease of access, only cells containing forest tracks were used. In the field, fine scale placement was chosen based on the presence of woodland. Cells that did not contain trees were excluded. Hair tubes were placed at distances of between 0 and 200 m from the nearest forest track (in increments of 50 m) with approximately the same number of tubes at each distance within a forest. A combination of Hawbakers marten lure (F&T Fur Harvester’s Trading Post, 10681 Bushey Road, Alpena, MI 49707), peanut butter and bread were used as attractants as these have previously proven effective (Chandrasekhar 2005; Roche 2008; Burki et al. 2009). Details of hair tube construction can be found in Online Resource 2.

Hair tube placement and scat transects in (a) Abernethy NNR, (b) Mar, (c) Inshriach and (d) Darnaway. The grey dots are hair tubes, placed at approximate density of 1 km−1. Dashed lines are transects used for scat collection and are placed along vehicle tracks. Transects were surveyed by walking up one side of the track and down the other, hence checking each track twice per survey
Scats were collected from Abernethy during May 2011 (Fig. 1). Scats were cleared 24 h prior to the first survey, and then two surveys were conducted on consecutive days so that all scats were ≤24 h old. All of the encountered scats were collected, essentially re-clearing transects of scats for subsequent collection rounds and enabling the time since defecation to be established, where the day of collection was ‘day zero’. Twenty-two scats were collected in individual pots and labelled with a unique identifier, then frozen at −20 °C within 8 h before transfer to a −80 °C freezer. In order to test the effect of exposure to rainfall and time since defecation on DNA quality, scats were thawed and a small section taken for DNA extraction (day zero samples). The remainder of the scat was split into two equal sections and allocated to one of two treatment groups. Samples in treatment one (exposed) were placed directly on a woodland floor in the University of Stirling grounds to replicate the conditions in which they were found. Samples in treatment two (sheltered) were placed in the same location, but raised off the ground and covered by a waterproof canopy. To test the effect of time since defecation (hereafter ‘time’), a small section of each scat was taken from both treatments at intervals of 2, 5, 9, 12 and 16 days.
Genetic analysis
Hair samples were removed from sticky pads with xylene. Extractions were performed using an adapted chelex-100 method (Walsh et al. 1991); a 1-cm root-section of hair was placed in 200 μl chelex (5 %), 7 μl dithiothreitol (DDT) and 1 μl proteinase K and agitated at 56 °C for approx. 5 h, centrifuged for 3 min and the supernatant incubated at 95 °C for 10 min. DNA was stored at −20 °C until required. The number of hair follicles in each extraction was recorded. Sex typing was performed using a 5′ nuclease TaqMan assay developed by Mullins et al. (2009) and real-time PCR using 5 μl Precision Master Mix (Primer Designs), 0.2 mM of either MMX or MMY forward and reverse primers and probes (MMX and MMY probe sequences are reversed from the text provided in Mullins et al. 2009 and are as follows: MMX, 5′-VIC-CCTGGTCTGAAAACT-MGB-3′ and MMY 5′-6FAM-TGTGTCTCTCTCTGTCAAMGB-3′) and 3 μl DNA template in a total volume of 10 μl. Amplification of ZFX (MMX) only signifies female DNA, whereas amplification of both ZFX and ZFY (MMY) signifies male DNA (Mullins et al. 2009). The PCR conditions were 2 min at 50 °C, 10 min at 95 °C, then 50 cycles of 15 s at 95 °C and 1 min at 60 °C. Two replicate amplifications were performed for each primer/probe. For real-time product detection, Ct value (i.e. the number of PCR cycles needed to obtain the required quantity of DNA) was recorded at a ΔRn threshold of 0.2.
For scat samples, genomic DNA was immediately extracted from day 0 samples using the QIAamp DNA stool mini kit (Qiagen, #51504) with a negative control. To avoid contamination, extractions were performed in an area of the laboratory reserved for DNA extraction. To test DNA amplification failure and error rates, two microsatellite loci were amplified (Mar08, Mar43; Natali et al. 2010) in one multiplex reaction of 10 μl containing 0.4 μM forward and reverse primers, 5 μl Qiagen Type-it PCR mastermix, 1 μl Q solution and 2 μl DNA template. After initial denaturation at 95 °C for 5 min, 40 cycles of 95 °C for 30 s, 63 °C for 90 s and 72 °C for 30 s were used followed by a final extension step of 60 °C for 30 min. Fragment analysis was performed at DNA Sequencing and Services (University of Dundee, Scotland, DD1 5EH) with negative and positive controls. Samples were scored using GeneMarker (Version 2.4.0) and verified by eye. Consensus genotypes were obtained for day 0 samples following the comparative multi-tubes approach (Frantz et al. 2003); each sample was initially amplified twice, then further replications were performed until a consensus was reached. Samples without a consensus after seven amplifications were discarded. Samples from each treatment and time period were extracted and amplified twice then compared to the consensus to quantify error rates, with a negative and positive control in each plate.
Statistical analysis
Darnaway was excluded from all analyses due to lack of hair samples. Visitation rate to hair tubes was analysed using a Generalised Linear Mixed effects Model (GLMM) with a binomial error distribution. The response variable was recorded as ‘visit’ or ‘no visit’ for each hair tube, replicated per session. To allow us to specifically test the effect of time on the rate of visitation, we included session as a proxy for time elapsed as a fixed covariate, as well as distance (question 1). Forest was included as a fixed factor and two-way interaction terms between distance and forests, and distance and session were included (question 2). Hair tube ID was included as a random factor. To test the effect of these variables on the sex ratio of visitors, the same analysis was used, but with the proportion of males as the response variable restricting analyses to samples with a positive sex ID only (question 2).
To determine whether PCR success for pine marten sex typing is affected by the number of hair follicles used in the extraction process, we calculated the mean Ct value per sample over positive rtPCR replicates. As the ZFX region is present in male and female pine martens and a Ct value is only obtained for positive samples, we included positive amplifications using the MMX locus only. There are, however, two copies of the ZFX region in female DNA for every one copy in male DNA, so it may take fewer cycles to obtain the threshold level of DNA template for female samples than for males; the effect of this bias should, however, be negligible as Ct value is unlikely to be reduced by more than one for females as compared to males. A Generalised Linear Model (GLM) with poisson error distribution was used with Ct value as the response variable and number of hairs as the explanatory variable (question 3).
Genotyping errors per amplification were categorised as allelic dropout (p), false alleles (f) and failure as described in Murphy et al. (2007), relative to the consensus genotype for each sample. Overall error rates were calculated using equations from Broquet et al. (2004):
Where p and f are the probability of allelic dropout and false alleles, respectively, at locus j. L refers to each scat within the treatment block, and A j and A hetj are the number of positive amplifications and the number of positive heterozygous amplifications, respectively, for the scat at locus j. D j and F j are the number of amplifications at locus j containing an allelic dropout and a false allele, respectively (Broquet et al. 2004).
The effects of time and exposure to rainfall on error rates (allelic dropout, false alleles and failure) were analysed using three GLMMs with a binomial (logit) distribution using proportional data from two repeated amplifications per sample for each combination of treatment, time and locus (question 4). As treatment commenced on day 2, samples from day 0 were not subject to the treatment conditions and so were not included in the models. Only successful samples (i.e. those that produced DNA) were included in the models for false alleles and allelic dropout. Treatment (exposed, sheltered) and locus were included as fixed factors, time (days) as a fixed covariate and an interaction between time and treatment included in all models. To account for pseudo-replication of scat samples, scat and ‘scat-half’ (i.e. the division of each scat between the two treatments) were included as random effects, with scat-half nested within scat.
For all analyses, we present estimates of the full model to avoid bias associated with stepwise deletion of non-significant terms (Whittingham et al. 2006). We present likelihood ratio test results for the deletion of each interaction term from the full model or each main effect from a model with main effects only (Faraway 2005; Zuur et al. 2009). Prediction uncertainty of the full models is calculated using N = 1000 random draws from the estimated parameter distributions and presented as the 95 % quantiles of the resulting distributions (Gelman and HIll 2007; Zuur et al. 2009). Analyses were performed in R version 3.1.0 (R core team 2014).
Results
Overall, hair samples were obtained on 20 % of occasions (115 samples, 572 tube nights; Online resource 1). Of the 115 samples, 69 (60 %) provided a positive sex type, with 23 samples from males and 46 from females.
Hair tube placement
Visitation rate varied over time, with a higher predicted visitation rate as sessions progressed from one (0.07; 0.01–0.10) to four (0.18; 0.13–0.25; Fig. 2), but did not significantly affect the sex ratio of visitors. Neither the distance of the tube from the nearest track nor the identity of the forest significantly improved model fit for hair tube visitation rate or the sex ratio of visitors (Table 1).

Visitation rate to hair tubes by pine marten in Scotland. Data points represent predicted visitation rate from the GLMM (Table 1) and error bars represent the 95 % confidence intervals for the model from repeated model simulations using random draws from the estimated parameter distributions (Gelman and Hill 2007). The ‘forest’ parameter was set for Inshriach. The ‘distance’ parameter was set to its median value
Hair sex-typing success
The number of hair follicles used for DNA extraction had a significant effect on the number of PCR cycles needed to obtain the required quantity of DNA, as measured by Ct value (χ 2 = −2.08, df = 61, p = 0.036). As the number of hair follicles increased from 1 to >13, the Ct value decreased by 13 % (Fig. 3).

Ct value obtained from rtPCR of the ZFX region of each pine marten hair sample plotted against the amount of hair used in the extraction process. Data points are for observed data, solid lines represent predicted Ct value from the GLM and dashed lines represent the 95 % confidence intervals for the model prediction from repeated model simulations using random draws from the estimated parameter distributions (Gelman and Hill 2007)
Scat genotyping success
For the experimental study, a consensus genotype was established for 28 of 44 sample loci (22 samples, two loci). DNA amplification was successful in 63 % (421/666) of attempts over all loci, treatments and time periods. The average temperature for the duration of the study was 15 °C (7.7–23.7 °C), with 21.6 mm rainfall overall (University of Stirling weather station).
PCR failure
Time, treatment and locus all significantly affected failure rate (Table 2). Failure rate increased from 0.28 (0.18–0.43) at day 2 to 0.65 (0.48–0.79) at day 16 for exposed samples, but did not change significantly for sheltered samples: 0.22 (0.13–0.35) at day 2 to 0.29 (0.15–0.42) at day 16 (Fig. 4). Locus also improved model fit with the average failure rate over all treatments and time periods being higher for locus m08 than locus m43, with proportions of 0.58 (0.51–0.61) and 0.44 (0.37–0.51), respectively.

Failure rate of PCR amplifications with increasing sample age, for samples exposed to rainfall (black line) and those under shelter (grey line). Data points are for observed data, solid lines represent predicted failure rates from the GLMM (Table 2) and dashed lines represent the 95 % confidence intervals for the model prediction calculated from repeated model simulations using random draws from the estimated parameter distributions (Gelman and Hill 2007). The ‘locus’ parameter was set to locus m43
Allelic dropout and false alleles
For successful amplifications, overall rates of allelic dropout and false alleles were 0.25 and 0.33, respectively. Neither treatment, time nor genetic locus significantly improved model fit for allelic dropout (Table 2). The rate of false alleles increased with time for exposed samples only, from 0.19 (0.10–0.38) to 0.52 (0.28–0.78; Table 2; Fig. 5). Samples amplified using locus m08 contained false alleles in 0.47 (0.37–0.58) of cases, compared to 0.30 (0.21–0.41) of cases for samples amplified with locus m43.

Rate of occurrence of false alleles with increasing sample age, for exposed (black line) and sheltered (grey line) samples. Data points are for observed data, solid lines represent predicted failure rates from the GLMM (Table 2) and dashed lines represent the 95 % confidence intervals for the model from repeated model simulations using random draws from the estimated parameter distributions (Gelman and Hill 2007). The ‘locus’ parameter was set to locus m43
Discussion
We tested temporal and spatial hair tube use by pine marten populations in Scotland and assessed the impacts of time and exposure to rainfall on scat DNA quality. Hair tube visitation rates increased over time with, on average, 2.6× as many samples collected in the fourth session compared to the first. This increase supports previous findings in Ireland, where sampling success increased with time when hair tubes were checked every 4 to 6 weeks for 6 months (O’Mahony et al. 2012). The shorter time period of the current study means that this effect is not due to increased population density, but suggests the influence of two factors: an increase in the likelihood of different animals locating hair tubes over time; and the habituation of individual pine martens to particular hair tubes. The latter of these factors is less apparent as, of 15 identified individuals that made multiple visits during the study period, only one individual used a single hair tube for all of their visits (Kubasiewicz et al., unpublished data).
The proximity of hair tubes to forest tracks did not affect the overall visitation rate, or the sex ratio of visitors, suggesting that tubes along tracks are not avoided by either sex and that surveying along tracks does not bias the sample towards more males than females. There is no evidence to suggest that placing hair tubes on the edge of forest tracks favours certain individuals; of the 15 pine martens that visited more than one hair tube, 14 (93 %) visited tubes at multiple distances (Kubasiewicz et al., unpublished data). Placing hair tubes directly next to forest tracks reduces sampling effort, potentially allowing more samples to be collected per session or more sessions to be conducted.
The amount of amplifiable DNA obtained from hair samples is significantly increased by including more hair follicles in each reaction. Previous studies suggest that one hair is sufficient for accurate genotyping (Scheppers et al. 2007). Our analysis, however, suggests that including more follicles (up to 13) reduces PCR failure rates. Where funding, or time, prevent processing of all samples, researchers should favour samples with the most follicles to increase PCR success. However, as hair tubes do not prevent visitation by more than one animal per session, the risk that including more than one hair per reaction may produce erroneous genotypes (i.e. via contamination from the second visitor) must be considered. During a larger scale study of pine marten population density in Scotland, including 136–320 hair tube nights per forest, no erroneous genotypes were detected (Kubasiewicz et al., unpublished data). We cannot rule out the possibility that more than one individual was present in a sample, with different homozygous genotypes at one or more loci (i.e. this would present as a heterozygous genotypes which we would not recognise as erroneous). However, as pine martens are attracted to hair tubes with bait, which is removed once a visit has occurred, the chance of multiple visits is low.
Both time since defecation and the level of exposure affected DNA amplification, reinforcing previous findings of the importance of these factors. An increase in PCR failure occurred with time up to 16 days after deposition, but only for scats that were exposed to rainfall. Although this effect was also seen for false alleles, allelic dropout did not increase significantly with time or treatment. As only a small number of repeat amplifications were performed, the increased failure rate over time could have masked any decrease in quality i.e. fewer successful amplifications were available for errors to occur in. Nevertheless, our results highlight the interacting effects of time and rainfall on pine marten scats and we would encourage other studies to assess the drivers of DNA degradation in faecal samples from other mammals. The finding that PCR failure increases with time for scats exposed to rainfall, as opposed to error rates alone, indicates that studies which require identification only at the species level, as well as those requiring accurate individual identification, need to minimise the effect of these factors for ensure a cost- and time-effective strategy.
There was a significant difference in DNA quality and amplification success between the two loci tested. During initial planning of a project, we would recommend testing a range of potential microsatellite loci for relative success and error rates so that the most effective panel can be chosen. This should be considered as essential as optimising sample collection and storage conditions in developing an efficient and cost-effective process.
For scat collection, researchers must strike a balance between leaving sufficient time for samples to accumulate and collecting samples before DNA degrades, particularly during periods of rainfall. If longer sampling sessions are required where populations are thought to be at low density, genotyping success may be improved by sampling during drier periods. For hair, samples are usually collected from stationary sources such as hair tubes. As such, the time between sampling sessions must also take into account sample independence. For pine martens in Scotland, our data suggest that sessions of longer than 4 days are required to achieve this independence. Compared to hair samples, scats are relatively easy to collect in large numbers, making this a preferable method of data collection for large scale studies. Scat samples, however, are difficult to genotype due to high levels of genotyping error associated with the poor quality DNA recovered (Lucchini et al. 2002). It may be beneficial for future studies to evaluate the use of single nucleotide polymorphisms (SNPs), which are more successful for degraded samples (Fabbri et al. 2012). Sample quality, however, can be maximised by using as many hair follicles as possible per sample in the DNA extraction process. Sampling efficiency can also be improved by placing hair tubes on the edge of forest tracks to improve access by surveyors. Given the high rate of error associated with non-invasive genetic sampling, refinement of the process and consideration of environmental conditions associated with each species is paramount to making the process efficient and cost effective. This study provides guidance for improvements to non-invasive surveys of pine martens in Scotland and also highlights key areas for assessment prior to surveys of other mammalian species.
References
Boulanger J, Proctor M, Himmer S, Stenhouse G, Paetkau D, Cranston J (2006) An empirical test of DNA mark-recapture sampling strategies for grizzly bears. Ursus 17:149–158
Brinkman TJ, Schwartz MK, Person DK, Pilgrim KL, Hundertmark KJ (2010) Effects of time and rainfall on PCR success using DNA extracted from deer fecal pellets. Conserv Genet 11:1547–1552
Broquet T, Menard N, Petit E (2007) Noninvasive population genetics: a review of sample source, diet, fragment length and microsatellite motif effects on amplification success and genotyping error rates. Conserv Genet 8:249–260
Broquet T, Petit E (2004) Quantifying genotyping errors in noninvasive population genetics. Mol Ecol 13:3601–3608
Brøseth H, Flagstad Ø, Wärdig C, Johansson M, Ellegren H (2010) Large-scale noninvasive genetic monitoring of wolverines using scats reveals density dependent adult survival. Biol Conserv 143:113–120
Burki S, Roth T, Robin K, Weber D (2009) Lure sticks as a method to detect pine martens Martes martes. Acta Theriol 55:223–230
Caryl FM, Raynor R, Quine CP, Park KJ (2012a) The seasonal diet of British pine marten determined from genetically identified scats. J Zool 288:252–259
Caryl FM, Quine CP, Park KJ (2012b) Martens in the matrix: the importance of nonforested habitats for forest carnivores in fragmented landscapes. J Mammal 93:464–474
Chandrasekhar A (2005) The evaluation of bait-marking as a method for delineating pine marten (Martes martes) territories in South-east Ireland, Dissertation, University of Kent
Cossios ED, Madrid A, Condori JL, Fajard U (2007) Update on the distribution of the Andean cat Oreaflurus jacobita and the pampas cat Lynchailurus colocolo in Peru. Endanger Species Res 3:313–320
Croose E, Birks JDS, Schofield HW (2013) Expansion zone survey of pine marten (Martes martes) distribution in Scotland. In: Scottish Natural Heritage Commissioned Report No. 520
Davies GM, Legg CJ (2008) The effect of traditional management burning on lichen diversity. Appl Veg Sci 11:529–538
Fabbri E, Caniglia R, Mucci N, Thomsen HP, Krag K, Pertoldi C, Loeschcke V, Randi E (2012) Comparison of single nucleotide polymorphisms and microsatellites in non-invasive genetic monitoring of a wolf population. Arch Biol Sci 64:321–335
Faraway JJ (2005) Extending the Linear Model with R. Chapman & Hall, Boca Raton, FL
Frantz AC, Pope LC, Carpenter PJ, Roper TJ, Wilson GJ, Delahay RJ et al (2003) Reliable microsatellite genotyping of the Eurasian badger (Meles meles) using faecal DNA. Mol Ecol 12:1649–1661
Gelman A, Hill J (2007) Data analysis using regression and multilevel/hierarchical models. Cambridge University Press, New York
Gibbs JP, Snell HL, Causton CE (1999) Effective monitoring for adaptive wildlife management: lessons from the Galapagos Islands. J Wildl Manag 63:1055–1065
Head JS, Boesch C, Robbins MM, Rabanal LI, Makaga L, Kuhl HS (2013) Effective sociodemographic population assessment of elusive species in ecology and conservation management. Ecol Evol 3:2903–2916
Kohn MH, York EC, Kamradt DA, Haught G, Sauvajot RM, Wayne RK (1999) Estimating population size by genotyping faeces. Proc Royal Soc B Biol Sci 266:657–663
Lockie JD (1964) Distribution and fluctuations of the pine marten, Martes martes (L.), in Scotland. J Animal Ecol 33:8
Lucchini V, Fabbri E, Marucco F, Ricci S, Boitani l, Randi E (2002) Noninvasive molecular tracking of colonizing wolf (Canis lupus) packs in the western Italian Alps. Mol Ecol 11
Lynch ÁB, Brown MJF, Rochford JM (2006) Fur snagging as a method of evaluating the presence and abundance of a small carnivore, the pine marten (Martes martes). J Zool 270:330–339
Mullins J, Statham MJ, Roche T, Turner PD, O’Reilly C (2009) Remotely plucked hair genotyping: a reliable and non-invasive method for censusing pine marten (Martes martes, L. 1758) populations. Eur J Wildl Res 56:443–453
Murphy MA, Kendall KC, Robinson A, Waits LP (2007) The impact of time and field conditions on brown bear (Ursus arctos) faecal DNA amplification. Conserv Genet 8:1219–1224
Natali C, Banchi E, Ciofi C, Manzo E, Bartolommei P, Cozzolino R (2010) Characterization of 13 polymorphic microsatellite loci in the European pine marten Martes martes. Conserv Genet Resour 2:397–399
Nsubuga AM, Robbins MM, Roeder AD, Morin PA, Boesch C, Vigilant L (2004) Factors affecting the amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol Ecol 13:2089–2094
O’Mahony D, Turner P, O’Reilly C (2012) Population status of pine marten in an isolated refuge: the Mourne Mountains. In: A report to the Peoples Trust for Endangered Species and Northern Ireland Environment Agency
Panasci M, Ballard WB, Breck S, Rodriguez D, Densmore LD, Wester DB et al (2011) Evaluation of fecal DNA preservation techniques and effects of sample age and diet on genotyping success. J Wildl Manag 75:1616–1624
Piggott MP, Taylor AC (2003) Extensive evaluation of faecal preservation and DNA extraction methods in Australian native and introduced species. Aust J Zool 51:15
Roche T (2008) The use of baited hair traps and genetic analysis to determine the presence of Pine marten, Dissertation, Waterford Institute of technology
Scheppers TLJ, Frantz AC, Schaul M, Engel E, Breyne P, Schley L, Roper TJ (2007). Estimating social group size of Eurasian badgers Meles meles by genotyping remotely plucked single hairs. Wildl Biol 13:195–207
Sheehy E, Lawton C (2014) Population crash in an invasive species following the recovery of a native predator: the case of the American grey squirrel and the European pine marten in Ireland. Biodivers Conserv 23:753–774
Summers RW, Willi J, Selvidge J (2009) Capercaillie Tetrao urogallus Nest Loss and Attendance at Abernethy Forest, Scotland. Wildl Biol 15:319–327
Summers R, Dugan D, Proctor R (2010) Numbers and breeding success of capercaillies Tetrao urogallus and black grouse T. tetrix at Abernethy Forest, Scotland. Bird Stud 57:437–446
Taberlet P, Luikart G (1999) Non-invasive genetic sampling and individual identification. Mol Genet Anim Ecol 41-55
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N et al (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24
Twiddle CL, Quine CP (2011) Revealing the vegetation history of Inshriach Forest: application of new quantitative reconstruction techniques to pollen records covering 3000 years. Scott For 65:19–27
Uno R, Kondo M, Yuasa T, Yamauchi K, Tsuruga H, Tamate HB, Yoneda M (2012) Assessment of genotyping accuracy in a non-invasive DNA-based population survey of Asiatic black bears (Ursus thibetanus): lessons from a large-scale pilot study in Iwate prefecture, northern Japan. Popul Ecol 54:509–519
Walsh PS, Metzger DA, Higuchi R (1991) Chelex-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Weckel M, Giuliano W, Silver S (2006) Jaguar (Panthera onca) feeding ecology: distribution of predator and prey through time and space. J Zool 270:25–30
Whittingham MJ, Stephens PA, Bradbury RB, Freckleton RP (2006) Why do we still use stepwise modelling in ecology and behaviour? J Anim Ecol 75:1182–1189
Wilson DJ, Efford MG, Brown SJ, Williamson JF, McElrea GJ (2007) Estimating density of ship rats in New Zealand forests by capture-mark-recapture trapping. N Z J Ecol 31:47–59
Zuur A, Leno EN, Walker N, Saveliev AA, Smith GM (2009) Mixed effects models and extensions in ecology with R. Springer, New York
Acknowledgments
We would like to thank David Bavin, Lizzie Croose, Tara Curry, Melissa Simmons and Kayleigh McCrory for help with data collection; Stuart A’Hara, Bridget Laue (Forest Research) and Catherine O’Reilly (Waterford Institute of Technology, Ireland) for advice on genetic analysis; and Ron Summers (RSPB) for advice and support. This project was funded by the University of Stirling, Forestry Commission, Forest Research, the Royal Society of the Protection of Birds and Scottish Natural Heritage.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Andrzej Zalewski
Rights and permissions
About this article

Cite this article
Kubasiewicz, L.M., Minderman, J., Woodall, L.C. et al. Fur and faeces: an experimental assessment of non-invasive DNA sampling for the European pine marten. Mamm Res 61, 299–307 (2016). https://doi.org/10.1007/s13364-016-0276-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-016-0276-y