
Abstract
Non-invasive genetics is a powerful tool in wildlife research and monitoring, especially when dealing with elusive and rare species such as the Eurasian otter (Lutra lutra). Nevertheless, otter DNA obtained from scats and anal secretions appears to be exposed to very quick degradation processes, and the success rate in DNA amplification is lower than in other carnivores. We collected 191 samples from April to September 2011 along the river Sangro basin (Italy) which was recently re-colonized by the Eurasian otter. Using two sets of microsatellite loci (six Lut and seven OT loci), we investigated the influence of sample type and age, collection time, storage time, temperature and humidity on genotyping success and amplification success. We also tested the efficacy of different DNA extraction kits and storage buffer mediums. Finally, we compared amplification success rate, allelic dropout and false allele rates for each locus. We obtained a mean amplification success rate of 79.0 % and a genotyping success rate of 35.1 %. Fresh pure jellies yielded the highest amplification success and genotyping rate. Six microsatellite loci should be theoretically sufficient to distinguish the individual unrelated otters (PID = 0.001), while 13 loci were needed to distinguish sibling otters (PIDsibs = 0.002) in our population. We identified 11 otters, and molecular sexing ascertained the presence of five males, four females and two uncertain individuals. Generalized linear models highlighted a significant influence of sample type and age, temperature and humidity both on genotyping and amplification success.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The successful conservation and management of threatened wildlife requires accurate information on population abundance, structure and genetic variability. This kind of data can be difficult to obtain, especially when dealing with rare and elusive species. Advances in molecular ecology and data analysis offer accurate tools to study and monitor wild populations in a non-invasive way (Koelewijn et al. 2010; Arandjelovic et al. 2011; Caniglia et al. 2012; Brzeski et al. 2013; Pennell et al. 2013; Silveira Trinca et al. 2013).
After a strong decline during the 1980s, the Eurasian otter (Lutra lutra) was recovered in many European countries, and it has been recently downgraded from the ‘Vulnerable’ to the ‘Near Threatened’ category in the IUCN Red List (Ruiz-Olmo et al. 2008). In Italy, the species is considered rare, and it is still listed as ‘Endangered’ in the national Red List (Rondinini et al. 2013). The Eurasian otter disappeared from northern Italy during the 1980s (Macdonald and Mason 1983; Cassola 1986; Macdonald and Mason 1994), and its decline continued in the central and southern regions all over the 1990s while the other European populations were already recovering (Conroy and Chanin 2000). By 1990, its distribution was confined to the south with a small geographically and genetically isolated population (Spagnesi et al. 2000; Randi et al. 2003). The current otter’s presence in Italy consists of two subpopulations: a larger core in the south (Campania, Puglia, Basilicata and Calabria regions) and a small nucleus in the Abruzzo and Molise regions (south-central Italy) (Loy et al. 2004, 2010; Prigioni et al. 2007; Panzacchi et al. 2010). Based on the non-invasive genetic sampling (NGS) data, the otter population in Italy was estimated at 229–257 individuals (Prigioni et al. 2006). According to recent unpublished data, the south-central remnant population is now expanding northward, but the lack of detailed information about its structure and biology represents a serious impediment to the adoption of appropriate management actions and conservation plans. As direct observations are almost unfeasible due to the nocturnal habit of the species, NGS represents a valuable opportunity to get precious data on the status and structure of local populations. This is particularly important in Italy where the Eurasian otter shows the highest genetic differentiation among the European populations. This indicates that local adaptations might have developed there, and thus, Italian populations should be considered as an evolutionary significant unit (ESU) and should also be of special conservation interest due to their uniqueness (Randi et al. 2003; Mucci et al. 2010). The Eurasian otter is a suitable species for applying NGS due to the typical scat marking behaviour used for intra-specific communication (Chanin 1985; Gorman and Trowbridge 1989; Kruuk 2006). Eurasian otters use two main types of scent marking signs: spraints (faeces) and anal jellies (Kruuk 2006). Spraints consist mostly of food remains, while jellies are mucous secretions produced by their characteristic anal glands. Both spraints and jellies can be used for genetic analysis with different amplification success rates (Fike et al. 2004; Hájková et al. 2006; Lampa et al. 2008; Hájková et al. 2009). The main challenge in NGS studies concerns the reliability of results due to the high levels of genotyping errors and the low levels of genotyping success (Buchan et al. 2005; Waits and Paetkau 2005; Hájková et al. 2009). Many factors can influence genotyping success rates including sample type (Coxon et al. 1999; Hájková et al. 2006; Lampa et al. 2008), environmental conditions (Nsubuga et al. 2004), storage time and mediums (Frantz et al. 2003; Lampa et al. 2008), diet (Murphy et al. 2003) and DNA extraction and amplification protocols (Lampa et al. 2008). Non-invasive samples, especially faecal samples, usually contain a low quality and quantity of target DNA and high amounts of PCR inhibitors and non-target DNA resulting in low genotyping success and high error rates. Two main types of genotyping errors are linked to NGS: allelic dropout (ADO), stochastic failure of one allele of heterozygote to amplify, and false allele (FA), an amplification artefact. In both cases, the result is a mistakenly identified genotype. Genotyping errors have to be quantified and incorporated into subsequent analysis for population studies (Bonin et al. 2004; McKelvey and Schwartz 2004; Hájková et al. 2006). In this context, L. lutra is one of the most problematic species as the DNA in scats is exposed to a very quick degradation process and the DNA amplification success rate is usually very low (Jansman et al. 2001; Dallas et al. 2003; Hung et al. 2004; Prigioni et al. 2006; Ferrando 2008; Lampa et al. 2008; Hájková et al. 2009).
Every step from sample collection to DNA amplification presents some critical aspects that can potentially influence genotyping success. Our aim is to test and compare different types of non-invasive samples, sample ages, storage times, storage buffer mediums, extraction kits and nuclear microsatellite loci in order to optimize NGS protocols and choose the most appropriate microsatellite loci panel for genotyping wild Eurasian otters.
Materials and methods
Study area
The study has been conducted in the Sangro basin in central Italy (Fig. 1) which was recently re-colonized by the Eurasian otter after the species was declared extinct in the region during the 1980s (Cassola 1986). This area represents the northern boundary of the otter range in Italy. The Sangro River flows for about 122 km from 1,441 m slm to the Adriatic Sea, and the basin covers 1,545 km2. Spraints and anal jellies were collected along the Sangro River and its main tributaries. Marking sites were detected during a pilot study (from May to October 2010; Lerone et al. 2011) through linear transect sampling run according to the European standard survey method (Reuther et al. 2000).

Sampling sites and sites providing successfully genotyped individual otters in the Sangro river basin
Sample collection and storage
Sampling season extended from April to September 2011. Almost no otter signs were found under bridges or on river banks, while most of the marking sites were located on rocks in the middle of the water course. Despite the fact that temperature could be a critical issue for DNA degradation (Hájková et al. 2006), we decided to collect samples during the warm period as most of the marking sites were unreachable during winter. The exposure of samples to UV light, wind, rain, etc. was highly variable among sites. Before each sampling, old spraints and jellies were removed from each site. Sites were then checked daily until fresh samples were collected within 24 h of deposition. According to the estimated age, the samples were categorized as (i) ‘very fresh’—the moist samples collected within 24 h from deposition (48), (ii) ‘fresh’—the samples (deposed within 24 h or not) that appeared moist below the surface (13) and (iii) ‘medium/old’—the samples with uncertain deposition that did not appeared completely dry (5). The medium/old samples were collected at less responsive marking sites. The sampling started early in the morning and went on until the afternoon with the collection time recorded along with the mean daily temperature and the humidity as recorded by the nearest meteorological station in the region (Abruzzo). Samples were classified as spraint, jelly or mixed spraint (jelly in addition to prey remains; Hájková et al. 2009) and immediately stored in ethanol 96 % in 1.5 ml tubes in an ice box and later preserved at −20 °C (during the same day). A subset of 30 samples was divided in two halves with one half stored in ethanol 96 % and the other in white cell lysis buffer (WCLB) in order to test for the effect of the conservation buffer medium on PCR amplification success.
Also, the time period between sample collection and DNA extraction, defined as storage time, can influence PCR amplification success (Frantzen et al. 1998; Murphy et al. 2007; Lampa et al. 2008), and it was recorded and analyzed.
DNA extraction
As Lampa et al. (2008) reported a significant difference in the amplification success rate between two different extraction methods (Chelex® 100 and QIAmp® DNA Stool Mini Kit), DNA from 13 non-invasive samples was extracted according to the manufacturer protocols of two different kits, DNeasy Blood and Tissue Kit (Qiagen®) and ZR Genomic DNA II Kit (Zymo Research®). The 13 samples were divided into two parts each, and subsequently, each of the two parts was extracted by a different kit. The successive samples were only extracted with the better performing kit (DNeasy Blood and Tissue Kit, Qiagen®). Negative controls were included in each extraction to monitor contamination.
DNA amplification and genotyping
We analyzed otter samples with two sets of nuclear microsatellite loci. In particular, we genotyped six loci ‘Lut’ (set1: Lut453, Lut604, Lut701, Lut832, Lut833 and Lut902; Dallas et al. 1998) already used in many previous studies (Dallas et al. 2002; Kalz et al. 2006; Hájková et al. 2009) and seven loci ‘OT’ (set2: OT04, OT05, OT07, OT14, OT17, OT19, OT22; Huang et al. 2005) only recently used for L. lutra (Koelewijn et al. 2010; Quaglietta et al. 2013), but never explicitly compared to Lut loci. Due to the low genetic variability of our small otter population, we had to use both sets of loci for individual recognition. We defined successfully genotyped samples as those where all the 13 microsatellite loci revealed complete genotype. We estimated the theoretical probability of identity (PID), which is the probability that two individuals randomly sampled from a population share the same genotype, by using GeneAlEx v.6 (Peakall and Smouse 2006). All samples were initially screened for three microsatellite loci (OT07, OT14 and OT17) with four independent repeats per locus. Due to the high cost of analysis and the low quality of non-invasive samples, only samples with 50 % or more positive screening PCRs (Peatkau 2003) were further amplified four times at each of the remaining ten microsatellite loci and at ZFX/ZFY loci for genetic sexing (Mucci and Randi 2007). All samples which provided less than 50 % of positive screening PCRs were ‘negative’, and all the others were finally successfully genotyped and considered ‘positive’. Negative controls were always included to monitor for contaminations. PCRs were performed in 10-μl volumes using 2 μl of DNA solution, 1 μl of PCR buffer 10× (1.5 mM of MgCl2, 5 PRIME-Eppendorf), 1 μl of bovine serum albumin 0.2 % (BSA), 0.4 μl of 2.5 mM dNTP mix, 0.15 μl of each primer (10 μM), 0.25 units of Taq (5 PRIME-Eppendorf) and 5.25 μl of RNA-free water. In order to optimize the costs and the time spent for analyses, we ran five multiplex PCR (M1 included Lut453, OT05 and OT22; M2 included Lut833 and OT19; M3 included Lut604 and Lut 701; M4 included Lut832 and Lut902; and M5 included OT17 and OT07) and one simplex PCR for OT04. Microsatellite loci analyzed within each multiplex PCR were chosen on the basis of their allelic range using Multiplex Manager Software v.1.0 (Holleley and Geerts 2009) and then tested on tissue samples before using non-invasive samples. We decided to perform simplex PCRs for OT04 due to its low amplification rate in multiplex conditions. When further amplifications were needed to verify data, only simplex PCR was used. Amplifications were performed using the following thermal profile: a first denaturating step at 94 °C for 2 min, 45 cycles at 94 °C for 40 s, 55 °C for 60 s, 72 °C for 90 s and a final extension step at 72 °C for 10 min. PCR products were electrophoresed with GeneScan™-350 ROX (Applied Biosystem) as marker ladder in an ABI 3130XL sequencer, and allele sizes were scored using the software GENEMAPPER v.4.0® (Applied Biosystems). Consensus genotypes were reconstructed using the GIMLET software v.1.3.3 (Valiere 2002) and accepted heterozygotes only if the two alleles were seen in at least two replicates and homozygotes only if the same homozygous genotype was seen at least in four replicates. Uncertain loci were additionally replicated another four times with a simplex PCR protocol. False allele and allelic dropout rates were estimated using GIMLET software v.1.3.3, and allele frequencies and unique genotypes were identified by GenAlEx v.6.
Data analysis
We compared the two subset of samples stored in different buffer mediums and extracted by different kits by using the McNemar test for paired samples. We carried out a multifactor analysis on the whole data set to test the parameters that could affect genotyping success and PCR success rate, i.e. sample type (jelly, spraint and mixed spraint), sample age (very fresh, fresh and medium), collection time (h), mean daily temperature (°C) and humidity (%) and storage time (days). Generalized linear models (GLMs) were run on both the whole sample (successfully and unsuccessfully genotyped samples) and on the subset of successful samples by using the genotyping success (positive-negative) and the PCR success rate as the response variable respectively. Despite the fact that the PCR success rate should have a more informative value, we also used the genotyping success as a dependent variable to analyze the entire data set in order to predict significant factors to be evaluated during sampling. Multivariate models were compared using the Akaike information criterion (AIC) with the lowest value indicating the best fitting model. Statistical analyses were performed using the free software R v.3.0.2.
Results
Sample collection, storage and DNA extraction
A total of 191 samples were collected including spraints (n = 137), jellies (n = 29) and mixed spraints (n = 25). Of these, 113 were very fresh, 45 fresh and 32 medium/old samples. Storage time ranged from 3 to 24 days. Out of the 30 samples stored in two different storage mediums, 14 (46.7 %) samples stored in ethanol and 6 (20.0 %) stored in WCLB provided complete genotypes. Despite the difference being poorly significant, the results of McNemar’s chi-squared test for paired data (X 2 = 3.68, p = 0.05) indicates ethanol 96 % as a slightly better storage medium for our samples. Nevertheless, this result will need further confirmation since our small sample size (n = 30) could hide both a stronger difference and an equivalence between the two buffers. Only samples stored in ethanol 96 % were included in GLM analysis.
Despite having a genotyping success for the samples extracted with DNeasy Blood and Tissue Kit (Qiagen®) and ZR Genomic DNA II Kit (Zymo Research®) of 53.84 and 15.38 % respectively, statistical testing did not show any significant difference between the two kits (McNemar’s chi-squared test for paired data X 2 = 0.5, p = 0.48), which is likely due to the limited sample size (n = 13). However, we decided to continue the DNA extraction with the better performing Blood and Tissue Kit (Qiagen®). Only samples extracted by Qiagen kit were included in GLM analysis.
DNA amplification and genotyping
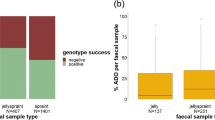
We performed a total of 7 158 PCR amplifications, and we completely genotyped 35.1 % (n = 67) of the total samples. The highest genotyping success was obtained from pure jellies (79.3 %) followed by mixed spraints (28.0 %) and spraints (27.0 %). We also obtained significant differences in the genotyping success among samples of different age with very fresh samples providing the highest success rate (42.5 %) followed by fresh (28.9 %) and medium/old (18.7 %) samples (Table 1).
All loci, but one (Lut453), were polymorphic, allele number ranged from one to four and the size of alleles ranged from 120 to 227 bp (Table 2). The OT loci were more variable than Lut loci as we identified a mean number of alleles per locus of 2.57 and 2.17 respectively (Table 2). In order to compare the performance of the 13 single loci, the mean values of PCR success rate and the ADO rates were reported in Table 3. The ADO rate differed significantly among different sample types (Kruskal-Wallis test: H = 11.4, p = 0.003). Jelly was the sample type with the lowest ADO rate.
As the allele size may influence the amplification success and ADO rates (Buchan et al. 2005; Broquet et al. 2007), we analyzed the effect of the mean molecular weight of DNA fragments on PCR success, ADO and FA rates. No relationship was found between fragment weight and allelic dropout rate (t = 1.26, p = 0.16) or false allele rate (t = −1.32, p = 0.21) while PCR success rate decreased significantly with increased median allele size (r 2 = 0.88, p = 1.4e-06).
Factors affecting genotyping and PCR success rate—multifactor analysis
Considering the genotyping success as the binary response variable (0 negative, 1 positive), the GLM multifactor analysis ran on the whole data set (positive and negative samples) showed that sample type and age as well as collection time and temperature significantly influence the genotyping success (Table 4). Jellies had a higher genotyping success followed by mixed spraints and spraints, while the genotyping success decreased with increasing mean daily temperature. Sample age and collection time had a lower influence on the response variable; nevertheless, the genotyping success was higher for very fresh samples collected earlier during the day. Storage time and mean daily humidity were not significant factors.
Considering the PCR success rate as the response variable, the GLMs performed on the successfully genotyped data set highlighted a significant influence of sample type and age as well as mean daily temperature and humidity (Table 5). For the genotyping success, the PCR success rate was higher for jellies than for mixed spraints and spraints. Amplification success was also higher for very fresh samples. The PCR success rate significantly decreased with increasing mean daily humidity, while it increased with high temperatures. The collection time had no significant effect on the genotyping success.
Individual identification
We analyzed the theoretical probability of identity among unrelated (PID) or sibling individuals (PIDsibs) (Fig. 2). Six microsatellite loci (Lut701, OT22, OT 17, OT19, Lut 833 and OT04) should be theoretically sufficient to distinguish the individual unrelated otters (PID = 0.001), while 13 loci were needed to distinguish sibling otters (PIDsibs = 0.002) with high probability (>99 %). All loci except OT17 resulted in Hardy-Weinberg equilibrium after the Bonferroni correction, and only nine of a total of 78 comparisons between loci resulted in linkage disequilibrium.

Probability of identity for unrelated individuals (PI) and for siblings (PIsibs) based on allele frequencies. The PI values by locus are reported on the right
Results showed the presence of at least 11 individual otters in the sampling area with a number of captures per individual ranging from 1 to 23. All individuals had a complete genotype (13 microsatellites) and were recognized as homozygotes at a particular locus if at least four independent PCR gave a coherent result. Molecular sexing was carried out according to the protocol of Mucci and Randi (2007). False allele errors for ZFX/ZFY sequences (119 and 178 bp) did not occur, while we found a mean ADO rate of 0.337 and 77 % of positive PCRs. We ascertained the presence of five males and four females and two individuals with uncertain molecular sexing for which only two successful PCR indicated homozygosity (females), which was not enough to accept the result as definite.
Discussion
Although our sampling extended during the warm period and almost no otter signs were found under bridges or on river banks causing our samples to become really exposed to environmental conditions, we succeeded in obtaining good results to characterize the otter population in the study area. The genotyping success on non-invasive samples in our study was 35.1 % which is a value falling within the range of prior studies on river otters (Dallas et al. 2003; Hung et al. 2004; Kalz et al. 2006; Prigioni et al. 2006; Arrendal et al. 2007; Ferrando et al. 2008; Lanszki et al. 2008; Hájková et al. 2009; Mowry et al. 2011). Jellies collected within 24 h from deposition had the highest genotyping success rate (79.3 %) and was consistent with previous studies which also reported a higher genotyping success for anal jelly samples (Coxon et al. 1999; Lampa et al. 2008; Hájková et al. 2009; Mowry et al. 2011). We estimated the rates of the main genotyping errors, allelic dropouts and false alleles in order to avoid unreliable results (Kalz et al. 2006; Hájková et al. 2009; Spiering et al. 2009, Bjǒrklund 2010; Guertin et al. 2012). The mean frequencies of ADO and FA among samples in our study were 14.4 and 0.3 %, respectively, with ADO rates being lower than that found in previous studies (Ferrando et al. 2008; Lampa et al. 2008; Hájková et al. 2009). Despite the extraction methods possibly influencing the genotyping success (Lampa et al. 2008), we did not find any significant differences between extractions performed with DNeasy Blood and Tissue Kit (Qiagen®) and ZR Genomic DNA II Kit (Zymo Research®). A marginally significant difference was found among the two compared storage mediums with the ethanol performing better than WCLB. Nevertheless, this topic needs further investigation since our small sample size could have influenced the result.
Besides sample type and age, the most important factors affecting the genotyping success were the mean daily temperature and the collection time. Increasing temperatures significantly reduced the genotyping success in our study, and Nsubuga et al. (2004) also recorded a decreasing DNA amount with increasing temperature in mountain gorilla scat samples. While Hájková et al. (2006) reported a lower PCR success rate for samples collected at high temperatures, we had a positive effect on PCR success. This result appears in contrast with the effect of temperature on the genotyping success and with what was expected since the high temperatures should cause an increase in hydrolytic enzyme activity (Hájková et al. 2006). It could be possible that temperature effect was confounded with other undetected environmental factors.
In accordance with Hájková et al. (2006), we recorded no effect of storage time on genotyping success. The range of storage times was too narrow to test the effect on PCR success rate. Lampa et al. (2008) observed a drastic decrease in amplification success (from 80 to 63 %) after 1 week, but in our data set, only a few samples exceed 7 days of storage.
We found that humidity negatively influenced the PCR success rate, as reported for other carnivores (Farrell et al. 2000; Murphy et al. 2007) and was expected for the increased activity of enzyme and bacteria in moist conditions (Lampa et al. 2008). As for genotyping success, jelly samples presented the highest PCR success rate. The high PCR success of jellies compared with mixed spraints seems to support the distinction made by Lampa et al. (2008) between the two types of mucous secretions found in jellies and in ‘spraints plus mucus’.
We analyzed seven Lut and six OT microsatellite loci in order to highlight the better performing microsatellites. In small and isolated otter population, we studied had a low genetic variability, and the number of alleles per locus was lower than in other studies on both L. lutra and Lontra canadensis (Hájková et al. 2009; Brzeski et al. 2013; Cohen et al. 2013). OT19 had the highest number of alleles (4) while Lut453 was monomorphic.
In accordance with Buchan et al. (2005), we detected an influence of DNA fragment size on genotyping success. On the contrary, we did not observe any increase in the ADO and FA rate with increasing fragment size.
Direct observation or radiotracking of rare and elusive species is difficult and expensive, but the need for demographic and ecological data is critical for monitoring and conserving wild populations. The traditional spraint survey method used to estimate abundance in Eurasian otter populations (Reuther et al. 2000) is quickly being improved by the addition of non-invasive genetic data. Non-invasive genetics has the potential to provide more detailed data about population abundance, structure, sex ratio, spatial organization and degree of kinship, but there is also the need to increase the success rate and the reliability of information that can be obtained by this approach.
In conclusion, we found the sample type and age, air temperature and humidity and collection time as important factors to account for when planning a non-invasive genetic sampling of Eurasian otter wild populations. Nevertheless, previous studies (Lampa et al. 2008; Bonesi et al. 2012) highlighted also other important factors such as time to extraction, sex and dominance factors. Further investigations of environmental factors, DNA analysis protocols and otter marking behaviour may provide valuable improvements in non-invasive genetic studies. The development of new microsatellite primers could greatly improve the individual identification in small and little variable populations which are the most urgent target of conservation biology.
References
Arandjelovic M, Head J, Rabanal LI, Schubert G, Mettke E, Boesch C, Robbins M, Vigilant L (2011) Non-invasive genetic monitoring of wild central chimpanzees. PLoS ONE 6(3)
Arrendal J, Vilà C, Björklund M (2007) Reliability of non invasive genetic census of otters compared to field censuses. Conserv Genet 8:1097–1107
Bjǒrklund M (2010) A method for adjusting allele frequencies in the case of microsatellite allele dropout. Molec Ecol Notes 5:676–679
Bonesi L, Hale M, Macdonald DW (2012) Lessons from the use of non-invasive genetic sampling as a way to estimate Eurasian otter population size and sex ratio. Acta Theriol 58(2):157–168
Bonin A, Bellamain E, Eidesen PB et al (2004) How to track and assess genotyping errors in population genetic studies. Mol Ecol 13:3261–3273
Broquet T, Ménard N, Petit E (2007) Noninvasive population genetics: a review of sample sources, diet, fragment length and microsatellite motif effect on amplification success and genotyping error rates. Conserv Genet 8:249–260
Brzeski KF, Szykman Gunther M, Black JM (2013) Evaluating river otter demography using noninvasive genetic methods. J Wildlife Manage 77(8):1523–1531
Buchan JC, Archie EA, Van Horn RC, Moss CJ, Alberts SC (2005) Locus effects and sources of error in noninvasive genotyping. Mol Ecol 5:680–683
Caniglia R, Fabbri E, Cubaynes S, Gimenez O, Lebreton JD, Rande E (2012) An improved procedure to estimate wolf abundance using non-invasive genetic sampling and capture-recapture mixture models. Conserv Genet 13:53–64
Cassola F (1986) La lontra in Italia. Censimento, distribuzione e problemi di conservazione di una specie minacciata. In: The Otter in Italy. Survey, distribution e conservation of an endangered species. WWF Italia, Serie Atti e Studi n.5, Roma, 135 pp
Chanin P (1985) The natural history of otters. Facts on File Inc., New York
Cohen TM, Narkiss T, Dolev A, Ben-Ari Y, Kronfeld-Schor N, Guter A, Saltz D, Bar-Gal GK (2013) Genetic diversity of the Eurasian otter (Lutra lutra) population in Israel
Conroy JWH, Chanin PF (2000) The status of the Eurasian otter (Lutra lutra) in Europe—a review. In: Conroy JWH, Yoxon P, Gutleb AC (eds.) Proceedings of the first otter toxicology conference, Isle of Skye. September 2000. Journal of the International Otter Survival Fund No 1:7–28
Coxon K, Chanin P, Dallas J, Sykes T (1999) The use of DNA fingerprinting to study population dynamics of otters (Lutra lutra) in Southern Britain: a feasibility study. R&D Technical Report W202, Environment Agency, Bristol, United Kingdom
Dallas JF, Piertney SB (1998) Microsatellite primers for Eurasian otter. Mol Ecol 7:1247–1251
Dallas JF, Marshall F, Piertney SB, Bacon PJ, Racey PA (2002) Spatially restricted gene flow and reduced microsatellite polymorphism in the Eurasian otter Lutra lutra in Britain. Conserv Genet 3:15–29
Dallas JF, Coxon KE, Sykes T, Chanin PRF, Marshall F, Carss DN, Bacon PJ, Piertney SB, Racey PA (2003) Similar estimates of population genetic composition and sex ratio derived from carcasses and faeces of Eurasian otter Lutra lutra. Mol Ecol 12:275–282
Farrell LE, Roman J, Sunquist ME (2000) Dietary separation of sympatric carnivores identified by molecular analysis of scats. Mol Ecol 9:1583–1590
Ferrando A, Lecis R, Domingo-Roura X, Ponsà M (2008) Genetic diversity and individual identification of reintroduces otters (Lutra lutra) in north-eastern Spain by DNA genotyping of spraints. Conserv Genet 9:129–139
Fike JA, Serfass TL, Beheler AS, Rhodes OE Jr (2004) Genotyping error rates associated with alternative sources of DNA for the north American river otter. IUCN Otter Spec Group Bull 21A
Frantz AC, Pope LC, Carpenter PJ, Roper TJ, Wilson GJ, Delahay RJ, Burke T (2003) Reliable microsatellite genotyping of the Eurasian badger (Meles meles) using faecal DNA
Frantzen MAJ, Silk JB, Fergusen JWH, Wayne RK, Kohn MH (1998) Empirical evaluation of preservation methods for faecal DNA. Mol Ecol 7:1423–1428
Gorman ML, Trowbridge BJ (1989) The role of odor in the social lives of carnivores. In: Git-tleman JL (ed) Carnivore behavior, ecology, and evolution. Cornell University, Ithaca, pp 57–88
Guertin DA, Ben-David M, Harestad AS, Elliott JE (2012) Fecal genotyping reveals demographic variation in river otters inhabiting a contaminated environment. J Wildlife Manage
Hájková P, Zemanova B, Bryja J, Hájek B, Roche K, Tkadlec E, Zima J (2006) Factors affecting success of PCR amplification of microsatellite loci from otter faeces. Mol Ecol Notes 6:559–562
Hájková P, Zemanová B, Roche K, Hájek B (2009) An evaluation of field and non invasive genetic methods for estimating Eurasian otter population size. Conserv Genet 10:1667–1681
Holleley CE, Geerts PG (2009) Multiplex Manager 1.0: a crossplatform computer program that plans and optimizes multiplex PCR. BioTechniques 46(7):511–517
Huang CC, Hsu YC, Lee LL, Li SH (2005) Isolation and characterization of tetramicrosatellite DNA markers in the Eurasian otter (Lutra lutra). Mol Ecol 5:314–316
Hung CM, Li SH, Lee LL (2004) Faecal DNA typing to determine the abundance and spatial organisation of otters (Lutra lutra) along two stream systems in Kinmen. Anim Conserv 7:301–311
Jansman HAH, Chanin PRF, Dallas JF (2001) Monitoring otter populations by DNA typing of spraints. IUCN Otter Spec Bull 18:12–19
Kalz B, Jewgenow K, Fickel J (2006) Structure of an otter (Lutra lutra) population in Germany—results of DNA and hormone analyses from faecal samples. Mamm Biol 71(6):321–335
Koelewijn HP, Pérez-Haro M, Jansman HAH, Boerwinkel MC, Bovenschen J, Lammertsma DR, Niewold FJJ, Kuiters AT (2010) The reintroduction of the Eurasian otter (Lutra lutra) into the Netherlands: hidden life revealed by noninvasive genetic monitoring. Conserv Genet 11:601–614
Kruuk H (2006) Otters ecology, behaviour and conservation, 2nd edn. Oxford University, Oxford
Lampa S, Gruber B, Henle K, Hoehn M (2008) An optimization approach to increase DNA amplification success of otter faeces. Conserv Genet 9:201–210
Lanszki J, Hidas A, Szentes K, Révay T, Lehoczky I, Weiss S (2008) Relative spraint density and genetic structure of otter (Lutra lutra) along Drava River in Hungary. Mamm Biol 73:40–47
Lerone L, Mengoni C, Randi E, Carpaneto GM, and Loy A (2011) Non-invasive genetic sampling of Eurasian otter in its Italian northern range. IUCN-XIth International Otter Colloquium- Otters in a warming world (Pavia, Italy, 2011). Hystrix It J Mamm (n.s.), Suppl. 2011: 109
Loy A, Bucci L, Carranza ML, De Castro G, Di Marzio P, Reggiani G (2004) Survey and habitat evaluation for a peripherical population of the Eurasian otter in Italy. IUCN Otter Spec. Group Bull. 21A
Loy A, Boitani L, Bonesi L, Canu A, Di Croce A, Fiorentino PL, Genovesi P, Mattei L, Panzacchi M, Prigioni C, Randi E, Reggiani G (2010) The Italian action plan for the endangered Eurasian otter Lutra lutra. Hystrix It J Mamm (ns) 21(1):19–33
Macdonald SM, Mason CF (1983) The Otter Lutra lutra in Southern Italy. Biol Conserv 25(2):95–101
Macdonald SM, Mason CF (1994) Status and conservation need of the otter (Lutra lutra) in the western Paleartic. Nat Environment 67:1–54, Council of Europe Press
McKelvey KS, Schwartz MK (2004) Genetic errors associated with population estimation using non-invasive molecular tagging: problems and new solutions. J Wildlife Manage 68:439–448
Mowry RA, Gompper ME, Eggert LS (2011) River otter population size estimation using noninvasive latrine surveys. J Wildlife Manage 75(7):1625–1636
Mucci N, Randi E (2007) Sex identification of Eurasian otter (Lutra lutra) non-invasive DNA samples using ZFX/ZFY sequences. Conserv Genet 8:1479–1482
Mucci N, Arrendal J, Ansorge H, Bailey M, Bodner M, Delibes M, Ferrando A, Fournier P, Fournier C, Godoy JA, Hajkova P, Hauer S, Heggberget TM, Heidecke D, Kirjavainen H, Krueger HH, Kvaloy K, Lafontaine L, Lanszki J, Lemarchand C, Liukko UM, Loeschcke V, Ludwig G, Madsen AB, Mercier L, Ozolins J, Paunovic M, Pertoldi C, Piriz A, Prigioni C, Santos-Reis M, Sales Luis T, Stjernberg T, Schmid H, Suchentrunk F, Teubner J, Tornberg R, Zinke O, Randi E (2010) Genetic diversity and landscape genetic structure of otter (Lutra lutra) populations in Europe. Conserv Genet 11(2):583–599
Murphy MA, Waits LP, Kendall KC (2003) The influence of diet on faecal DNA amplification and sex identification in brown bears (Ursus arctos). Mol Ecol 12:2261–2265
Murphy MA, Kendall KC, Robinson A, Waits LP (2007) The impact of time and field conditions on brown bear (Ursus arctos) faecal DNA amplification. Conserv Genet 8:1219–1224
Nsubuga AM, Robbins MM, Roeder AD, Morin PA, Boesch C, Vigilant L (2004) Factors affecting the amount of genomic DNA extracted from ape faeces and the identification of an improved sample storage method. Mol Ecol 13:2089–2094
Panzacchi M, Genovesi P, Loy A (2010) Piano d’azione nazionale per la conservazione della lontra (Lutra lutra). Min. Ambiente - ISPRA
Paetkau D (2003) An empirical exploration of data quality in DNA-based population inventories. Mol Ecol 12:1375–1387
Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Ecology Notes 6:288–295
Pennell MW, Stansbury CR, Waits LP, Miller CR (2013) Capwire: an R package for estimating population census size from non-invasive genetic sampling. Mol Ecol Resour 13:154–157
Prigioni C, Remonti L, Balestrieri A, Sgrosso S, Priore G, Mucci N, Randi E (2006) Estimation of European otter (Lutra lutra) population size by fecal DNA typing in Southern Italy. J Mamm 87(5):855–858
Prigioni C, Balestrieri A, Remonti L (2007) Decline and recovery in otter Lutra lutra populations in Italy. Mamm Rev 37:71–79
Quaglietta L, Fonseca VC, Hájková P, Mira A, Boitani L (2013) Fine-scale population genetic structure and short-range sex-biased dispersal in a solitary carnivore, Lutra lutra. J Mammal 94(3):561–571
Randi E, Davoli F, Pierpaoli M, Pertoldi C, Madsen AB, Loeschcke V (2003) Genetic structure in otter (Lutra lutra) populations in Europe: implications for conservation. Anim Conserv 6:93–100
Reuther C, Dolch D, Green R, Jahrl J, Jefferies D, Krekemeyer A, Kucerova M, Madesn AB, Romanowsky J, Roche K, Ruiz-Olmo J, Teubner J, Trinidade A (2000) Surveying and monitoring distribution and population trends of the Eurasian otter (Lutra lutra): guidelines and evaluation of the standard method for surveys as recommended by the European Section of the IUCN/SSC Otter Specialist Group. Habitat 12
Rondinini C, Battistoni A, Peronace V, Teofili C (2013) Lista Rossa IUCN dei Vertebrati Italiani. Comitato Italiano IUCN e Ministero dell’Ambiente e dela Tutela del Territorio e del Mare, Roma
Ruiz-Olmo J, Loy A, Cianfrani C, Yoxon P, Yoxon G, de Silva PK, Roos A, Bisther M, Hájková P, Zemanová B (2008) Lutra lutra. In: IUCN 2009. IUCN Red List of Threatened Species. Version 2009.1. www.iucnredlist.org. Accessed 30 May 2014
Silveira Trinca C, Fernandes Jaeger C, Eizirik E (2013) Molecular ecology of the Neotropical otter (Lontra longicaudis): non-invasive sampling yields insights into local population dynamics. Biol J Linn Soc 109:932–948
Spagnesi M, Toso S, De Marinis AM (eds) (2000) I Mammiferi d’Italia, Istituto Nazionale per la Fauna Selvatica. Ozzano Emilia, Italy
Spiering PA, Gunther MS, Wildt DE, Somers MJ, Maldonado JE (2009) Sampling error in non-invasive genetic analyses of an endangered social carnivore. Conserv Genet 10:2005–2007
Valiere N (2002) GIMLET: a computer program for analysing genetic individual identification data. Mol Ecol Notes 2:377–379
Waits LP, Paetkau D (2005) Noninvasive genetic sampling for wildlife biologists: a review of applications and recommendations for accurate data collection. J Wildlife Manage 69(4):1419–1433
Acknowledgments
We thank Prof. C. Pertoldi and the anonymous reviewer for their precious advices which greatly improved the manuscript. We also thank A. Brambilla, A. Leone, L. Nelli, A. Palladini, R. Caniglia, F. Davoli, E. Fabbri, F. Mattucci and N. Mucci for their suggestions and constant support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Cino Pertoldi
Rights and permissions
About this article

Cite this article
Lerone, L., Mengoni, C., Carpaneto, G.M. et al. Procedures to genotype problematic non-invasive otter (Lutra lutra) samples. Acta Theriol 59, 511–520 (2014). https://doi.org/10.1007/s13364-014-0195-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-014-0195-8