
Abstract
Contrary to chicken and livestock mammals, duck genome has not been explored much. Nowadays a relatively small number of reports on molecular variability and mapping of loci in Peking ducks has been published. Therefore, the objective of this study was to detect single loci affecting body weight, carcass and meat traits in Peking ducks (Anas platyrhynchos). The study was based on an F2 cross between two parental lines A-55 and GL-30. Phenotypes of 387 birds from generation F2 including carcass and meat quality traits were collected. Linkage map, of the linkage group CAU1, consisting of 29 microsatellite markers was constructed. One highly significant (p < 0.01) QTL for skin weight was identified at the beginning (0 cM) of the linkage group CAU1. Two significant QTL (p < 0.05) were found for fatness of leg muscle and leg muscle electrical conductivity – 24 hours post slaughtering (EC24h) at 178 and 100 cM, respectively. Additionally one suggestive QTL for odour of leg muscle was identified at 265 cM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although over the last decades the duck population seems to have stabilised, duck meat plays an important role in the European poultry market. The number of slaughtered ducks in the structure of commercially slaughtered poultry has increased significantly and for several years has accounted for 4 %. Furthermore, waterfowl breeding is very important in other regions of the world, mainly in eastern and southern Asia (FAO database 2013). Hence, the breeders’ efforts have been focused on increasing carcass yield and reduction of fatness using classical polygenetic approaches as well as improving environmental conditions and processing management. However, current advances in molecular genetics have generated new opportunities for poultry genetic improvement programs due to association of chromosomal regions with performance traits (see e.g. Boschiero et al. 2013). The development of methods to characterise genetic variation at the DNA level has enabled mapping of loci that affect quantitative traits. There is a general agreement from theoretical and simulation studies that application of marker assisted selection (MAS) has the potential to increase the rate of genetic gain especially if traditional selection is less effective (Goddard and Hayes 2009).
Unfortunately, contrary to chicken as well as livestock mammals, duck genome is still not sufficiently known. Nowadays a relatively small number of reports on molecular variability and mapping of loci in Peking ducks has been published (Huang et al. 2005, 2006, 2008; Maak et al. 2000; Wang et al. 2011; Wu et al. 2008). Huang et al. (2006) developed a genetic linkage map for the duck. However, for reliable construction of the map, detection of many polymorphic DNA markers is necessary. In the case of ducks, the most saturated microsatellite marker map is located on chromosome 1. As mentioned above, a few papers have been published on detection of significant associations of some loci with production traits in ducks. For instance, Huang et al. (2007a, b), identified some loci affecting body weight, and conformation traits as well as carcass and meat quality traits in Beijing ducks. Also some molecular studies were performed on interspecific hybrid mule ducks by Kileh-Wais et al. (2013). They detected QTLs for quality of breast meat and fatty liver. It is important to refine the duck linkage map in order to construct a consensus map integrating knowledge of the duck genome from many studies. Inclusion of information from different populations should allow verification of marker positions and their respective positions on the linkage map.
The objective of this study was to describe molecular characteristics of the analysed loci, detect single loci affecting body weight, carcass and meat quality traits in Peking duck (Anas platyrhynchos).
Material and methods
Experimental population
The study was based on an F2 cross between two parental lines A-55 and GL-30. The A-55 line originates from Polish Peking, whereas the GL-30 line originates from French Peking. The birds were maintained at a duck farm in Nowa Wieś Wielka near Poznan (Poland). The experimental procedures were approved by the Local Ethical Commission for Animal Experiments in Poznan (Poland). Five males from A-55 line were crossed with 23 females from GL-30 line. From the first generation of offspring five males and 28 females were selected as parents of the next generation. The second generation of offspring (F2) consisted of 390 individuals out of which three were removed from the experiment.
Phenotypes
Birds were fed ad libitum from 0 to 6 weeks of age with a starter diet and then feed was restricted from 6 to 11 weeks of age with a grower diet. During the second period, individuals were kept in colony cages. More details on housing and feeding conditions of the experimental birds are given by Mucha et al. (2014). In the 11th week of life (before slaughtering), body weight was measured (using electronic balance AXIS B15S with ±5 grams accuracy) on life birds after 12 hours without access to feed and with permanent access to water. Slaughter and post slaughtering processing were performed in the same standard technological conditions. For good identification, during the whole process, an individual code was installed on each carcass. The carcasses were refrigerated for 24 hours at 4 °C. Dissection was performed using the method described by Ziołecki and Doruchowski (1989). Carcasses and their components were weighted by electronic balance RADWAG WPT 5C with ±0.2 gram accuracy. The following performance traits were measured on 387 animals from the F2 generation: body weight at 11 weeks of age (BW) [g], eviscerated carcass (with neck and giblets - without gizzard) weight (CW) [g], breast muscle weight including superficial and deep muscle (BMW) [g], leg muscles weight including thigh and shank (LMW) [g], neck weight without skin (NW) [g], wing weight (WW) [g], skin weight with subcutaneous fat (SW) [g], abdominal fat weight (AFW) [g], skeleton with the back muscle and inedible elements (SBM) [g], liver weight (LW) [g], hydrogen ion concentration (pH) in breast muscle - 15 minutes post slaughter (BMpH15), hydrogen ion concentration (pH) in breast muscle - 24 hours post slaughter (BMpH24), hydrogen ion concentration (pH) in leg muscle - 15 minutes post slaughter (LMpH15), hydrogen ion concentration (pH) in - leg muscle pH 24 hours post slaughter (LMpH24), electrical conductivity of breast muscle - 15 minutes post slaughter (BMEC15) [mS/cm], electrical conductivity of breast muscle - 24 hours post slaughter (BMEC24) [mS/cm], electrical conductivity of leg muscle - 15 minutes post slaughter (LMEC15) [mS/cm], electrical conductivity of leg muscle - 24 hours post slaughter (LMEC24) [mS/cm], water holding capacity of breast muscle (WHCBM) [mg%], water holding capacity of leg muscle (WHCLM) [mg%], thermal drip of breast muscle (TDBM), thermal drip of leg muscle (TDLM) [%], lightness (L*) of breast muscle (LBM) [%], lightness (L*) of leg muscle (LLM), colour of raw breast muscle (CRBM), colour of raw leg muscle (CRLM), odour of raw breast muscle (ORBM), odour of raw leg muscle (ORLM), fatness of raw breast muscle (FRBM), fatness of raw leg muscle (FRLM), general appearance of raw breast muscle (ARBM), general appearance of raw leg muscle (ARLM), total score of raw breast muscle (TSRBM), total score of raw leg muscle (TSRLM), water content of breast muscle (WCBM) [%], water content of leg muscle (WCLM) [%], lightness (L*) of both breast and leg muscles (after mincation and homogenisation) were evaluated using an automated Minolta Chroma Meter CR-310 (with light source D65 and 8 mm measuring cell). Concentration of hydrogen ions in muscle was measured using a portable Mettler-Toledo pH meter (with mercurous chloride electrode) whereas measurements of muscle electrical conductivity were performed by LF-STAR apparatus of the Matthus company. Water holding capacity was determined by the use of modified method of Grau and Hamm (Hamm 1986) whereas thermal leak was measured by a method given by Pikul (1993). Sensory evaluation (for CRBM, CRLM, ORBM, ORLM, FRBM, FRLM, ARBM, ARLM, TSRBM, TSRLM ) of muscles was conducted by a sensory panel (five trained persons) according to the Polish Standards using 4 point scale (2 – undesirable quality, 5 – desirable quality). Final score per single sample was expressed as arithmetic mean (±0.1 point).
Blood samples were collected from parents, as well as from the F1 and F2 offspring. Molecular identification of sex of F2 individuals was determined according to the procedure described by Clinton et al. (2001).
Genotyping
Markers were selected from the first linkage group CAU1 (Huang et al. 2006). In total 33 microsatellite markers were used out of which four had to be removed from further analyses due to problems with PCR amplification and gel electrophoresis. DNA from all animals was extracted from blood using standard methods. Blood was collected from clavicle vain into tubes with EDTA and stored in -20 °C. DNA was isolated from full blood. DNeasy Tissue from Qiagen was used for DNA extraction. DNA concentration was assessed by spectrometry and electrophoresis on 0.8 % agarose gel. The starters for amplification of microsatellite sequences were chosen based on literature (Buchholz et al. 1998; Huang et al. 2005; Maak et al. 2003). The loci were amplified using PCR with fluorescent tagged starters. Reactions were carried out in 4 loci multiplex with Type-it Microsatellite PCR Kit (Qiagen). Reaction was carried out in 10 μl volume which contained 5 μl 2x concentrated reaction mix Type-it, 1 μl of DNA matrix (approx. 50 ng) and each of the starters with 0.25 μM concentration. Amplification was done in 2720 Thermal Cycler (Applied Biosystems) with the program: an initial denaturation step at 95 °C for 5 min followed by 30 cycles of 95 °C for 30 s, 50-65 °C for 90 s, 72 °C for 30 s, and final extension step at 60 °C for 30 min. Products of amplification were diluted with water (10 to 100 times), 1 μl was added to 9 μl of formamid containing 0.5 μl DNA GeneScan-600 LIZ Size Standard (Applied Biosystems) and denatured for 5 min in 95 °C. Capillary electrophoresis was performed in ABI Prism 3130XL (Applied Biosystems), with 36 cm long capillaries, polymer POP7 and G5 filter. The allele sizes were read in Peak Scanner v 1.0 (2006, Applied Biosystems, http://www.appliedbiosystems.com). Molecular analyses were performed by the Laboratory of Molecular Biology Techniques at Faculty of Biology of Adam Mickiewicz University in Poznan (Poland).
Preliminary analysis
Basic statistics describing the data (averages, standard deviations as well as minimum and maximum values for consecutive traits) were estimated with R package (R Development Core Team 2010). Departures from normal distribution of the analysed traits were verified with the Shapiro-Wilk test.
Quantitative trait locus analyses
The genotype data were used to construct a linkage map with CRIMAP (Green et al. 1990). Quantitative trait locus analysis was performed under an F2 model using GRID-QTL software (Seaton et al. 2006). The experiment-wide 5 % and 1 % significance thresholds were calculated for each trait by a permutation analysis (5000 permutations per trait); 95 % confidence interval was calculated for significant QTL based on bootstrap analysis with 5000 iterations. Genome scans were performed using the forward selection approach at 1 cM intervals.
The following unitrait genetic additive model was used for QTL mapping:
where: y is a vector of phenotypes, β is a vector of fixed effects of sex, a is a vector of additive polygenic effects, q is a vector of additive QTL effect, e is a vector of residuals as well as X and Z are design matrices relating records to fixed and random effects, respectively.
Results and discussion
Linkage map
The initial linkage map was constructed out of 25 markers. Position of four markers was difficult to determine based on the available data. Two of them were added to the map in subsequent steps. Marker CAUD020 was positioned between CAUD131 and CAUD024 as this position had the highest likelihood and was in agreement with a previously reported map (Huang et al. 2006). Based on the same condition, CAUD074 was placed between CAUD112 and CAUD093. The remaining two markers (CAUD029 and CAUD086) could not be added to the linkage map because they had equally likely position at the beginning as at the end of the linkage map. Linkage map based on the current analysis does differ in certain areas form the map presented by Huang et al. (2006). Length of the linkage group CAU1 in the current study was 437.9 cM (Table 1), whereas in the previous study it spanned across 317 cM (Huang et al. 2006). This indicates a difference in recombination rates between the two populations. Moreover, in some regions the order of the markers was different. In total, seven markers appeared in different order in comparison to Huang et al. (2006).
Number of alleles in the analysed loci ranged between 2 (CAUD029, APH23, APH15, CAUD025, CAUD020) and 16 (CAUD024). Mean heterozygosity of all loci was 0.55. However, loci CAUD086 and CAUD018 had low heterozygosity of 0.19 and 0.15, respectively. Polymorphic information content (PIC) of the analysed markers ranged between 0.22 and 0.87 with a mean of 0.53 (Table 1).
Analysed traits
Means, coefficient of variation, minimum and maximum values for the analysed traits are presented in Table 2. Increased trait variability can be partially explained by crossbreeding effect. It should be recalled that all birds recorded were kept under the same environmental conditions.
Coefficients of variation were small for many of the meat quality traits such as pH (CV between 1.7 and 3.2), colour and odour of breast meat (CV of 2.2) or water content of breast meat (CV of 1.3 and 2.1, respectively). Low variability of those traits makes search for potential QTL more difficult.
QTL analysis
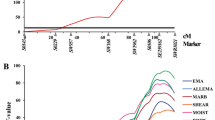
Current study was only limited to markers from linkage group CAU1 which limited the area of the duck genome that could be scanned for potential QTL. Estimated genetic additive effects and profiles of F statistics for significant QTL for three traits (FRCM, ORLH, LMpH24), mapped on linkage group CAU1, are presented in Fig. 1a-c.

a Estimated genetic additive effects and profile of F statistics for significant QTL for fatness of leg muscle mapped on linkage group CAU1. Dashed line represents 0.05 experiment-wide significance threshold, and the solid line marks 0.01 significance threshold b Estimated genetic additive effects and profile of F statistics for significant QTL for odour of leg muscle mapped on linkage group CAU1. Dashed line represents 0.05 experiment-wide significance threshold, and the solid line marks 0.01 significance threshold c Estimated genetic additive effects and profile of F statistics for significant QTL for electrical conductivity of leg muscle EC 24 hours mapped on linkage group CAU1. Dashed line represents 0.05 experiment-wide significance threshold, and the solid line marks 0.01 significance threshold
Two significant QTL (p < 0.05) were found for FRLM and LMEC24 at 141 and 63 cM, respectively. Additionally, a QTL for ORLM was identified at 228 cM. It has to be noted that confidence intervals for all of the identified QTL were large and ranged between 265 and 390 cM (Table 3). Current confidence intervals were much larger than those reported by Huang et al. (2007a, b), whose largest interval did not exceed 257 cM. Given a much larger sample size in the current study such high confidence intervals can be considered as unexpectedly high. Nowadays most projects focused on finding genes that influence economically important traits in livestock are conducted using dense SNP panels. Unfortunately, SNP chip for duck has not been released yet. Therefore researchers working with this species have to rely on older microsatellite markers. SNP markers could be used in a future analysis to fine map the identified QTL. Because of their high abundance they should allow for a much higher resolution of the QTL scan. SNPs are expected to be closer to the sites responsible for variation than microsatellites (Vignal et al. 2002). In principal the use of SNP markers can not only improve the localisation of the QTL but also estimates of their magnitude (Slate et al. 2009). Additionally SNP markers allow for a better targeting of a wide range of genomic regions such as exons, promoters, splice sites and other regulatory regions (Vignal et al. 2002).
The current study did not confirm the previously identified QTL for breast muscle weight (Huang et al. 2007a) and body weight (Huang et al. 2007b) located in linkage group CAU1. We obtained two peaks for body weight at 93 cM and 358 cM. However they did not reach the significance threshold. The second peak was in a similar region as the QTL flanked by markers CAUD131 and CAUD126 reported by Huang et al. (2007b). Due to map differences position of this QTL is different in the current study (358 cM) than in the results reported by Huang et al. (2007b), where the location was reported at 101 cM. In the analysis of breast muscle weight one peak was obtained at 358 cM (flanking markers: CAUD103 and CAUD025). This region was not in the neighbourhood of the QTL reported by Huang et al. (2007a). The three QTLs (FRLM, ORLM and LMEC24) identified in the current study have not been reported before. Therefore they can be considered as novel QTL for duck meat quality.
QTLs identified in the current study explained between 2.7 to 3.2 % of phenotypic variation. The three QTL explained a much smaller part of variance in comparison with the loci identified by Huang et al. (2007a, b), who reported values between 3.29 and 17.68 % for carcass weight and meat quality.
Conclusions
Three significant QTL for meat quality were found on duck linkage group CAU1. Confidence intervals for the identified QTL were large which requires further fine mapping analysis to obtain more precise location of causative genes. The three QTL identified in the current study have not been reported before. Therefore they can be considered as novel QTL for duck meat quality.
References
Boschiero C, Jorge EC, Ninov K, Nones K, Rosario MF, Coutinho LL, Ledur MC, Burt DW, Moura ASAMT (2013) Association of IGF1 and KDM5A polymorphisms with performance, fatness and carcass traits in chickens. J Appl Genet 54:103–112
Buchholz WG, Pearce JM, Pierson BJ, Scribner KT (1998) Dinucleotide repeat polymorphisms in waterfowl (family Anatidae): characterization of a sex-linked (Z-specific) and 14 autosomal loci. Anim Genet 29(4):323–325
Clinton M, Haines L, Belloir B, McBride D (2001) Sexing chick embryos: a rapid and simple protocol. Br Poult Sci 42:134–138
Goddard ME, Hayes BJ (2009) Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet 10:381–391
Green P, Falls K, Crooks S (1990) Document for CRI-MAP, version 2.4. Washington Univ, School of Medicine, St. Louis
Hamm R (1986) Functional properties of the myofibrillar system and their measurements. In: Bechtel PJ (ed) Muscle as Food. Academic Press, Orlando, p 144
Huang Y, Tu J, Cheng X, Tang B, Hu X, Liu Z, Feng J, Lou Y, Lin L, Xu K, Zhao Y, Li N (2005) Characterization of 35 novel microsatellite DNA markers from the duck (Anas platyrhynchos) genome and cross-amplification in other birds. Genet Sel Evol 37:455–472
Huang Y, Zhao Y, Haley C, Hu S, Hao J, Wu C, Li N (2006) A genetic and cytogenetic map for the duck (Anas platyrhynchos). Genet 173:287–296
Huang Y, Haley CS, Hu S, Hao J, Wu C, Li N (2007a) Detection of quantitative trait loci for body weights and conformation traits in Beijing ducks. Anim Genet 38:525–526
Huang Y, Haley CS, Wu F, Hu S, Hao J, Wu C, Li N (2007b) Genetic mapping of quantitative trait loci affecting carcass and meat quality traits in Beijing ducks (Anas platyrhynchos). Anim Genet 38:114–119
Huang Y, Li N, Burt DW, Wu F (2008) Genomic research and applications in the duck (Anas platyrhynchos). World’s Poult Sci J 64:329–341
Kileh-Wais M, Elsen JM, Vignal A, Feves K, Vignoles F, Fernandez X, Manse H, Davail S, Andre JM, Bastanelli D, Bonnal L, Filangi O, Baeza E, Guemene D, Genet C, Bernardet MD, Dubos F, Marie-Etancelin C (2013) Detection of QTL controlling metabolism, meat quality and liver quality traits of the over fed interspecific hybrid mule duck. J Anim Sci 91:588–604
Maak S, Wimmers K, Weigend S, Neumann K (2000) First seven microsatellites developed for the Peking duck (Anas platyrhynchos). Anim Genet 31:233
Maak S, Wimmers K, Weigend S, Neumann K (2003) Isolation and characterization of 18 microsatellites in the Peking duck (Anas platyrhynchos) and their application in other waterfowl species. Molec Ecol Not 3(2):224–227
Mucha S, Gornowicz E, Lisowski M, Grajewski B, Radziszewska J, Szwaczkowski T (2014) Genetic parameters of carcass traits in ducks from a crossbreed population. Ann Anim Sci 14:43–53
Pikul J (1993) Technological assessment of raw materials and products of poultry industry. Ann Agr Univ Poznań 63-64 (in Polish, with English summary)
PN-ISO 6658 (1998) Sensory analysis. Methodology. General guidance: 1-21 (in Polish)
R Development Core Team (2010) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Seaton G, Hernandez J, Grunchec JA, White I, Allen J, De Koning DJ, Wei W, Berry D, Haley C, Knott S (2006) GridQTL: A Grid Portal for QTL Mapping of Compute Intensive Datasets. Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, August 13-18, 2006. Belo Horizonte, Brazil
Slate J, Gratten J, Beraldi D, Stapley J, Hale M, Pemberton JM (2009) Gene mapping in the wild with SNPs: guidelines and future directions. Genetica 136:97–107
Vignal A, Milan D, Sancristobal M, Eggen A (2002) A review on SNP and other types of molecular markers and their use in animal genetics. Genet Sel Evol 34:275–305
Wang C, Li S, Yu W, Xin Q, Li C, Feng YPX, Gong Y (2011) Clonning and expression profiling of the VLDLR gene associated with egg performance in duck (Anas platyrhynchos). Genet Sel Evol 43:1–29
Wu F, Huang Y, Ma Y, Hu S, Hao J, Li N (2008) Evaluation of genetic diversity and relationships within and between two breeds of duck based on microsatellite markers. Progr Nat Sci 19:1581–1986
Ziołecki J, Doruchowski W (1989) Method of slaughter value evaluation in poultry. COBRD: 1-32 (in Polish)
Internet Pages
FAO database (2013) faostat3.fao.org (updated October 2, 2013).http://www.appliedbiosystems.com
Acknowledgement
The research project was supported by the Polish Ministry of Science and Higher Education, grant No N N311 239838. The authors are grateful to Dr. hab. Anna Wolc for her valuable comments. Two anonymous reviewers are thanked for constructive remarks and suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mucha, S., Grajewski, B., Gornowicz, E. et al. Mapping quantitative trait loci affecting some carcass and meat traits in duck (Anas platyrhynchos). J Appl Genetics 55, 497–503 (2014). https://doi.org/10.1007/s13353-014-0224-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-014-0224-y