
Abstract
Mirabegron is a potent and selective β3-adrenoceptor agonist developed for the treatment of overactive bladder. In vitro studies demonstrated that mirabegron partly acts as a (quasi-) irreversible, metabolism-dependent inhibitor of CYP2D6. The effect of steady-state mirabegron on single doses of the sensitive CYP2D6 substrates metoprolol (100 mg) and desipramine (50 mg) was assessed in two open-label, one-sequence crossover studies in healthy subjects (CYP2D6 extensive metabolizers). Mirabegron 160 mg/day increased metoprolol maximum plasma concentration (C max) 1.90-fold (90 % confidence interval [CI] 1.54; 2.33) and total exposure (AUC0-∞) 3.29-fold (90 % CI 2.70; 4.00) in 12 males (study 1). Mean metoprolol half-life increased from 2.96 to 4.11 h. α-Hydroxymetoprolol C max and AUC to last measurable concentration decreased 2.6-fold and 2.2-fold, respectively. In study 2, mirabegron 100 mg/day increased desipramine C max 1.79-fold (90 % CI 1.69; 1.90) and AUC0-∞ 3.41-fold (90 % CI 3.07; 3.80) in 14 males and 14 females. Mean desipramine half-life increased from 19.5 to 35.8 h. C max of 2-hydroxydesipramine decreased ~twofold, while AUC increased ~1.3-fold. Desipramine was administered again 2 weeks after the last mirabegron dose. Desipramine C max and AUC0-∞ were still ~1.13-fold increased; the 90 % CIs fell within the 0.80–1.25 interval. All treatments were well tolerated. In conclusion, mirabegron is a moderate CYP2D6 inhibitor (ratio and 90 % CI <5.0).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Mirabegron, a selective and potent human β3-adrenoceptor agonist (Takasu et al. 2007), is approved for the treatment of overactive bladder (OAB) in Japan, the United States and the European Union. The therapeutic dose is 50 mg once daily. Clinical studies conducted in Europe, Australia, and North America have shown that daily administration of mirabegron to patients with OAB for 12 weeks resulted in significant improvements in OAB symptoms when compared with placebo-treated controls (Khullar et al. 2013; Nitti et al. 2012).
Mirabegron is metabolized through multiple pathways including amide hydrolysis by esterases, direct uridine diphospho-glucuronosyl transferase (UGT)-mediated glucuronidation and—to a lesser extent—N-dealkylation or oxidation presumably by cytochrome P450 (CYP) (Takusagawa et al. 2012b). Hepatic oxidative metabolism is primarily mediated by CYP3A4, with a minor role for CYP2D6 both in vitro and in vivo (Takusagawa et al. 2012c; Lee et al. 2013). In vitro studies have demonstrated that mirabegron is an inhibitor of CYP2D6 and a weak inhibitor of CYP3A4 (Takusagawa et al. 2012c). The CYP2D6 inhibition was NADPH- (reduced nicotinamide adenine dinucleotide phosphate) and time-dependent, which was partly resistant to dilution, suggesting that mirabegron may partly act as an irreversible or quasi-irreversible, metabolism-dependent inhibitor of CYP2D6 (Takusagawa et al. 2012a).
CYP2D6 is involved to some extent in the metabolism of many drugs; it is important to evaluate the CYP2D6 inhibitory potential of mirabegron in humans. Therefore, the impact of mirabegron on the pharmacokinetics of the CYP2D6 substrates metoprolol and desipramine was assessed. In addition, the recovery of CYP2D6 activity was explored. The safety and tolerability of the treatments were also evaluated.
Metoprolol is a selective β1-adrenoceptor antagonist commonly used in the management of acute myocardial infarction, angina, hypertension, and cardiac arrhythmias. It is extensively metabolized into at least four metabolites by α-hydroxylation, O-demethylation and subsequent oxidation into metoprolol acid, and by deamination. Although the α-hydroxylation accounts for only 10 % of metoprolol metabolism, this pathway is exclusively mediated by CYP2D6 (McGourty et al. 1985; Lennard et al. 1986). Desipramine, a commonly used tricyclic antidepressant, is metabolized to 2-hydroxydesipramine almost exclusively mediated by CYP2D6 (Sallee and Pollock 1990), making it a sensitive and selective probe substrate for CYP2D6 inhibition. Both metoprolol and desipramine can be classified as sensitive substrates for CYP2D6 and have been recommended as probe drugs for assessing CYP2D6 metabolism during drug development (European Medicines Agency 2010; US Food and Drug Administration 2012).
Earlier in the development, mirabegron was used as immediate-release capsules, and this formulation was used in the metoprolol study. The oral controlled absorption system (OCAS) formulation was administered in the desipramine study. The OCAS tablet has a relative bioavailability of around 80 % compared with the immediate-release capsule (Sawamoto et al. 2011; van Gelderen et al. 2009). The maximum plasma concentration of the OCAS formulation is about 20 % lower than that of the immediate-release capsule, while it was attained about 1.5 h later after a single dose (data on file).
2 Methods
Two single-center, Phase I, drug–drug interaction studies in healthy subjects were conducted. Study 1 (protocol 178-CL-005) was performed at Pharma Bio-Research Group (now PRA International) (Zuidlaren, The Netherlands) and the study protocol was approved by an Independent Ethics Committee (“Stichting Beoordeling Ethiek Biomedisch Onderzoek”, Assen, The Netherlands). Study 2 (protocol 178-CL-058) was conducted at SGS-Aster (Paris, France), and the study protocol was approved by the “Comité de Protection des Personnes CPP Ile de France III” (Hôpital Tarnier-Cochin, Paris, France). The studies were conducted in accordance with the Declaration of Helsinki, the Good Clinical Practice guidelines of the International Conference on Harmonisation, and local laws and regulations. All participants gave written, informed consent before study entry, after full explanation of the study.
2.1 Participants
All participants were healthy adults aged 18–55 years (study 1: only males), geno- and phenotyped as extensive metabolizers (EMs) for CYP2D6, with a body mass index (BMI) ≤30 kg/m2 and body weight between 60 and 100 kg (study 1) or a BMI ≥18.5 and <30 kg/m2 (study 2). Eligible subjects were of good health (determined by physical examination, 12-lead ECG, vital signs, and clinical laboratory evaluations) and had no clinically significant medical history. Female subjects of child-bearing potential were required to use adequate non-hormonal contraception. Concomitant medication was not allowed, except for the incidental use of acetaminophen. All subjects were required to comply with study restrictions (e.g., diet, exercise, and alcohol).
2.2 Study designs
Study 1 was a open-label, one-sequence crossover study to evaluate the effect of multiple doses of mirabegron on the metabolism of metoprolol in 12 healthy male subjects. All subjects received a single oral dose of 100 mg metoprolol tartrate (Genfarma, The Netherlands) on Day 1, followed by a 1-day washout. Subsequently, all subjects were administered oral dose of mirabegron 160 mg immediate-release capsules (Astellas Pharma, former Yamanouchi, Leiden, The Netherlands) once daily for 5 days (Days 3–7), with a single dose of 100 mg metoprolol tartrate co-administered on Day 7. All doses were administered after overnight fasting. Metoprolol pharmacokinetics were determined with and without mirabegron by taking sequential blood samples up to 48 h after dosing on Days 1 and 7. Mirabegron pharmacokinetics were determined by collecting blood samples predose on Days 3–7, postdose up to 14 h on Day 6, and postdose up to 48 h after dosing on Day 7. Subjects remained in the study center from the day before the first dose until 48 h after the last dose.
Study 2 assessed the effect of multiple doses of mirabegron on the pharmacokinetics of a single dose of desipramine in healthy male (14) and female (14) subjects using the open-label, one-sequence crossover design. In period 1, subjects received a single oral dose of 50 mg desipramine as two tablets of 25 mg desipramine HCl (Norpramin®, Sanofi-Aventis, USA) on Day 1. On Days 5–23, subjects received daily oral doses of 100 mg mirabegron OCAS tablets (Astellas Pharma Technologies, Norman, Oklahoma, USA), with a single 50 mg dose of desipramine co-administered on Day 18. After a washout of 2 weeks, subjects received a final dose of 50 mg desipramine on Day 38 in period 2. Blood samples for mirabegron plasma concentrations were collected predose on Days 16, 17, 18, and 38 and up to 24 h after dosing on Day 18. Blood samples for desipramine and 2-hydroxydesipramine plasma concentrations were collected predose and up to 72 h after dosing on Days 1, 18, and 38; and 96 and 120 h after dosing on Day 18. All treatments were administered after a overnight fast. Subjects remained in the study center from the day before the first dose until Day 23 and from Day 37 until Day 41.
Safety assessments included physical examinations, vital signs, resting 12-lead ECGs, safety clinical laboratory tests (biochemistry, hematology, and urinalysis), and adverse event (AE) monitoring.
2.3 Genotyping and phenotyping
Prior to study inclusion, subjects were genotyped for the CYP2D6 *3, *4, *5, *6, and xN alleles using an extra-long polymerase chain reaction (XL-PCR)-based assay to amplify the entire coding region of the CYP2D6 gene. Subsequently, an Allele Specific PCR was executed to determine the wild-type or mutation versions of the CYP2D6*3 (SNP: rs35742686), CYP2D6*4 (SNP: rs3892097), or *6 allele (SNP: rs5030655) (Heim and Meyer 1990; Evert et al. 1994). The CYP2D6*5 allele was determined using an XL-PCR with a forward primer specific for exon 9 of the CYP2D7 gene and a reverse primer specific for the 3′ flanking region of the CYP2D6 gene (Johansson et al. 1996). CYP2D6 xN was determined using an XL-PCR with a forward primer specific for CYP2D6 and a reverse primer specific for the CYP2D6-CYP2D6 intergenic region (Steijns and Van Der Weide 1998). All PCR products were analyzed using agarose gel electrophoresis. Subjects who carried none of the five mutant alleles (CYP2D6 *3, *4, *5, * 6, or xN) were regarded as EMs. In study 2, validated real-time PCR-based assays were used. CYP2D6*3 was determined using a Pre-Developed Assay Reagents kit (Applied Biosystems, Foster City, CA, USA) specific for rs35742686. CYP2D6*4 (SNP: rs3892097) and CYP2D*6 (SNP: rs5030655) were determined using TaqMan® Drug Metabolism Assays from Applied Biosystems. CYP2D6*5 and xN were determined by a multiplex TaqMan® Gene Copy Number Assay (Applied Biosystems) using primers and a probe specific for exon 9 of the CYP2D6 gene. The RNase P gene was used as a reference gene using RNase P primers and probe. Subjects who carried none or one of the CYP2D6 *3, *4, *5, or *6 alleles were classified as intermediate metabolizers or EMs.
The expression of the genotype was confirmed at screening by dextromethorphan phenotyping. Urine was collected over 8 h after dextromethorphan intake (22 mg of dextromethorphan hydrobromide monohydrate as syrup in study 1 or 30 mg dextromethorphan hydrobromide as capsule in study 2). The urine samples were analyzed for dextromethorphan and dextrorphan using a validated bioanalytical method (Wieling et al. 2000). Urinary dextromethorphan/dextrorphan molar excretion ratio (metabolic ratio) was calculated for each subject. Subjects with a metabolic ratio <0.3 were classified as EMs for CYP2D6 (Schmid et al. 1985).
2.4 Sample analysis
Mirabegron was extracted from plasma by liquid–liquid extraction. The plasma samples (0.500 ml) for study 1 were analyzed by Analytico Medinet (now Eurofins Medinet, Breda, The Netherlands) and those (0.250 ml) of study 2 by PPD Development (Richmond, VA, USA). Mirabegron and internal standard were extracted from plasma by liquid–liquid extraction using hexane:ethylacetate 1:1 v/v (study 1) or methyl-t-butyl ether (study 2). YM-88796 was used as an internal standard in study 1 and YM-9661388 (d5-mirabegron) in study 2. The processed samples were analyzed by high-pressure liquid chromatography (HPLC) coupled with a tandem mass spectrometer (MS–MS) (study 1: MDS Sciex API 3000; study 2: MDS Sciex API 4000, Applied Biosystems) using positive ion mode atmospheric pressure chemical ionization (APCI) interface. The transitions monitored were m/z 397 → 260 for mirabegron, m/z 376 → 358 for YM-88796, and m/z 402 → 260 for YM-9661388. A method transfer validation demonstrated the compatibility of the methods used. Data support the accurate and precise quantification of mirabegron in study 1 over a concentration range of 1.00 [lower limit of quantification (LLOQ)] to 500 ng/ml plasma. Precision (% coefficient of variation [CV]) was <10.7 % and accuracy ranged from 94.4 to 108.0 %. For study 2, calibration ranged from 0.20 (LLOQ) to 100 ng/ml. Precision (%CV) was <5.3 % and accuracy ranged from 97.5 to 103.0 %.
Metoprolol and α-hydroxymetoprolol were analyzed by Cephac (now SGS Cephac, St Benoît Cedex, France). The analytes were extracted, together with the internal standard alprenolol, from plasma by liquid–liquid extraction using methylene chloride. The processed samples were analyzed by reversed phase (μBondapak C18) HPLC with fluorometric detection (λ excitation = 275 nm, λ emission = 300 nm). Calibration ranged from 5.00 (LLOQ) to 1,000 ng/ml plasma for both analytes. Precision (%CV) was <5.3 % for metoprolol and <5.0 % for α-hydroxymetoprolol, and accuracy ranged from 95.9 to 100.4 % and from 96.0 to 99.6 %, respectively. There was no interference from mirabegron.
Desipramine, 2-hydroxydesipramine and the internal standards, desipramine-d3 and 2-hydroxydesipramine-d3, respectively, were extracted from 100 μl of human plasma by liquid–liquid extraction by PPD Development (Middleton, WI, USA). The reconstituted samples were injected onto a Restek Pinnacle II Cyano column (50 × 2.1 mm, 5 μm). The samples were eluted with an isocratic mobile phase (63.8 % acetonitrile, 35.0 % methanol, 1.00 % water, 0.1 % of a 10 % ammonium formate solution, and 0.1 % of a 10 % formic acid solution). The analytes were monitored on a Sciex API 4000 with a detection mode of positive electrospray. Calibration ranged from 0.25 (LLOQ) to 100 ng/ml plasma. Precision (%CV) was <4.6 % for desipramine and <6.0 % for 2-hydroxydesipramine, and accuracy ranged from 99.3 to 103.7 % and from 102.9 to 106.4 %, respectively. There was no interference from mirabegron.
2.5 Pharmacokinetic analysis
Plasma and urine concentration data were analyzed by noncompartmental methods using WinNonlin version 3.1 or higher (Pharsight Corporation, Mountain View, CA, USA). The pharmacokinetic parameters included area under the concentration–time curve (AUC) up to the time of the last measurable concentration (AUC0-t), obtained by the linear-logarithmic trapezoidal method, AUC extrapolated to infinity (AUC0-∞), AUC over the dosing interval, maximum plasma concentration (C max), time to C max (t max), and half-life associated with the terminal slope (t 1/2). For desipramine, the AUC0-∞ ratio of metabolite over parent (ratio AUC0-∞ M/P) was calculated.
2.6 Statistical methods
2.6.1 Study 1
The within-subject CV for metoprolol was estimated to be 14.1 % for AUC0-∞, and 16.6 % for C max (Yuen et al. 2001). The power for the study, with 12 subjects, to yield 90 % confidence intervals (CIs) for the metoprolol AUC0-∞ and C max ratios in the presence/absence of mirabegron, which lie entirely within the specified no-effect range of 0.80–1.25, ranged from 88 % for a true ratio of 1.00–57 % for a ratio of 1.10.
Metoprolol AUC0-∞ and C max were the primary variables for the statistical analysis. AUC0-t for metoprolol, C max, and AUC0-t for α-hydroxymetoprolol were secondary. The variables were log-transformed and subjected to an ANOVA with subject and treatment as fixed effects. A 90 % CI was obtained for the difference of the true means (log-scale) for the combination treatment versus metoprolol alone, which was back-transformed to the original scale. No interaction would be concluded if the 90 % CIs for both AUC0-∞ and C max for metoprolol were contained within the 0.80–1.25 range.
2.6.2 Study 2
The power calculation assumed ratios for desipramine with or without mirabegron similar to those found for metoprolol in study 1 and aimed to show that mirabegron is not a strong inhibitor of CYP2D6, i.e., obtain the upper limits of 90 % CI for both the AUC and C max ratios below the 5.0 margin for strong inhibitors (European Medicines Agency 2010; US Food and Drug Administration 2012). The intrasubject CVs for desipramine AUC ranged from 12.4 to about 30 % and for C max from 11.4 to 22.1 % (Laine et al. 2004; Aarnoutse et al. 2005; Harris et al. 2007). If the intrasubject CV for AUC was not larger than 30 % and the true AUC ratio was close to 4.0 (and the true C max ratio close to 2.0), then a study with 23 evaluable subjects would have a power of about 80 % for achieving 90 % CIs for AUC and C max ratios, which both lie entirely below the 5.0 margin.
The same analysis as for study 1 was performed for desipramine AUC0-∞ and C max for the combination treatment versus desipramine alone on Day 1 (primary) and also for desipramine alone on Day 38 versus Day 1 (secondary, recovery of CYP2D6). In addition, the effect of sex was explored by calculating AUC and Cmax ratios with the same model separately for male and female subjects. No interaction would be concluded if the 90 % CIs for both AUC0-∞ and C max for desipramine were contained within the 0.80–1.25 range. If the 90 % CIs for the AUC and C max ratios both fell below the 5.0 margin then it would be concluded that mirabegron is not a strong inhibitor of CYP2D6. Pharmacokinetic parameters for 2-hydroxydesipramine are summarized with descriptive statistics only.
3 Results
3.1 Demographics and disposition
Subject disposition and demographic information for both studies are presented in Table 1. All randomized subjects received study medication and were included in the safety and pharmacokinetic analyses. All subjects in study 1 completed the study. One male subject in study 2 was discontinued on Day 36, in the washout between periods 1 and 2, due to the use of disallowed medication for the treatment of sinusitis.
Study 1 included only male subjects, while study 2 included both males and females. Apart from sex, which differed by study design, the demographics of the different groups were generally comparable.
3.2 Effect of mirabegron on metoprolol pharmacokinetics
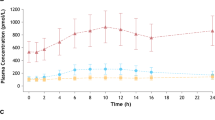
Figure 1 depicts the mean plasma concentration–time curves of metoprolol and α-hydroxymetoprolol after a single oral dose of 1000-mg metoprolol tartrate before and after multiple-dose mirabegron treatment (160 mg/day immediate release). Co-administration with mirabegron resulted in plasma concentrations of metoprolol being markedly increased, while those for α-hydroxymetoprolol decreased. The effect of mirabegron treatment on the pharmacokinetic parameters of metoprolol and α-hydroxymetoprolol is shown in Table 2. In all subjects, metoprolol and α-hydroxymetoprolol were quantifiable in a sufficient number of plasma samples to allow calculation of the pharmacokinetic parameters, except those for α-hydroxymetoprolol in the presence of mirabegron. Due to the fact that the α-hydroxymetoprolol plasma concentrations decreased in the presence of mirabegron, it was not possible to calculate t 1/2 and AUC0-∞. Mirabegron 160 mg/day increased metoprolol C max 1.90-fold and AUC0-∞ 3.29-fold. Mean t 1/2 increased from 2.96 to 4.11 h, while no effect on t max was observed. Because the 90 % CIs lay entirely above the no-effect ranges of 0.8–1.25, it was concluded that a drug–drug interaction occurred.

Mean plasma concentration versus time curves of metoprolol and its metabolite α-hydroxymetoprolol after a single dose of 100 mg metoprolol tartrate alone, and concurrently with steady-state mirabegron (160 mg once daily as immediate release capsules) in healthy subjects (n = 12) (study 1)
For α-hydroxymetoprolol, comparable t max values were observed in the absence and presence of mirabegron. The statistical analysis showed that C max decreased 2.6-fold and AUC0-t 2.2-fold in the presence of mirabegron.
During dosing from Days 3 to 7 the mean predose concentrations of mirabegron increased gradually. The mean ± standard deviation (SD) predose value on Day 7 (17.7 ± 4.5 ng/ml), however, was almost identical to the predose value on Day 6 (16.2 ± 4.3 ng/ml), indicating that steady state had been reached on the day of metoprolol co-administration. The mean ± SD C max of 297 ± 55 ng/ml was reached at a median of 2.25 h with an AUC over the dosing interval of 1,700 ± 270 ng.h/ml.
3.3 Effect of mirabegron on desipramine pharmacokinetics
The CYP2D6 inhibitory effect of multiple doses of mirabegron (OCAS tablet, 100 mg/day) was confirmed using another CYP2D6 probe drug, desipramine HCl at a dose of 50 mg. In addition, the recovery of CYP2D6 activity was assessed by a third dose of desipramine, 2 weeks after the last dose of mirabegron. Mean plasma concentrations of desipramine increased when co-administered with mirabegron, while those for the 2-hydroxy metabolite decreased almost twofold (Fig. 2). After the 2-week washout, the desipramine concentrations were only slightly higher than those after the first dose of desipramine alone, while the metabolite concentrations were similar. In the presence of mirabegron, a mean increase in desipramine C max and AUC0-∞ was observed (ratio 1.79 and 3.41, respectively) (Table 3), similar to those observed with metoprolol. After the 2-week washout, C max and AUC0-∞ were still about 1.13-fold increased, but remained within the range of 0.80–1.25. The t max for desipramine (6 h) was not affected by mirabegron.

Mean plasma concentration versus time curves of desipramine (a) and its metabolite 2-hydroxydesipramine (b) after a single dose of 25 mg desipramine HCl alone, and concurrently with steady-state mirabegron (100 mg once daily as oral controlled absorption system) in healthy subjects (n = 28, day 1 and 18; n = 27, day 38) (study 2)
In the presence of mirabegron, C max of 2-hydroxydesipramine decreased approximately twofold, while AUC0-∞ increased about 1.3-fold. Both parameters returned to values comparable with baseline after the 2-week washout. The mean ratio of the AUC0-∞ values of 2-hydroxydesipramine versus desipramine decreased from 0.942 on Day 1 to 0.372 on Day 18 (Table 3), showing that a relatively lower amount of desipramine was metabolized to 2-hydroxydesipramine. After the 2-week washout period, the metabolic capacity was recovered. The t 1/2 for both desipramine and 2-hyroxydesipramine almost doubled with mirabegron co-administration.
Throughout the study, desipramine and 2-hydroxydesipramine exposure were about twofold and 1.5-fold higher in female than in male subjects (Table 3); however, the effect of administration in the absence or presence of mirabegron was similar.
Assessment of predose trough concentrations of mirabegron indicated that steady state was reached before assessment of pharmacokinetics of desipramine in combination with mirabegron on Day 18. On Day 18, the mean ± SD C max of 162 ± 71 ng/ml for mirabegron was reached at a median of 5.00 h with an AUC over the dosing interval of 1,346 ± 461 ng.h/ml. On the day of the third desipramine dose, mirabegron plasma concentrations were still quantifiable in all subjects, with a mean value of 0.787 ng/ml. In general, mirabegron exposure was about 1.7-fold higher in female than in male subjects.
3.4 Safety
The treatments were well tolerated in both studies. There were no serious AEs or discontinuations due to an AE. In study 1, 26 treatment-emergent AEs were reported by 10 of 12 subjects. The most frequently reported were increased alanine aminotransferase (ALAT) (four subjects, all on Day 9 of the study, related to metoprolol and mirabegron), headache (four subjects, related to mirabegron and the co-treatment), and palpitations (three subjects, related to mirabegron). The increased ALAT levels were all less than twice the upper limit of normal and returned to normal at the post-study visit. In study 2, nine of 28 subjects reported 17 treatment-emergent AEs. Here, the most frequently reported AE was hard feces, which was reported by four subjects. All AEs had resolved at the end of the studies. Mirabegron did not affect the cardiovascular responses to metoprolol (i.e., decreased mean pulse rate and systolic blood pressure and—to a lesser extent—diastolic blood pressure) or desipramine (i.e., increased mean pulse rate and blood pressure).
4 Discussion
In vitro findings suggested that mirabegron partly acts as a (quasi-) irreversible, metabolism-dependent inhibitor of CYP2D6 and as such has potential to cause clinically relevant drug–drug interactions, although the in vivo C max to in vitro concentration with 50 % inhibition (IC50)/2 ratio was only 0.042 (Takusagawa et al. 2012a). Therefore, the effect of mirabegron on the pharmacokinetics of two probe drugs for CYP2D6, metoprolol and desipramine, both sensitive CYP2D6 substrates (European Medicines Agency 2010; US Food and Drug Administration 2012), was assessed. The mean C max and AUC0-∞ of a single dose of 100-mg metoprolol increased 1.90-fold and 3.29-fold, respectively, in the presence of steady-state mirabegron (160 mg/day immediate release) with an increase in metoprolol t 1/2 of 39 %. When a single dose of 50-mg desipramine was added to steady-state mirabegron (100 mg/day OCAS), mean C max and AUC0-∞ of desipramine increased by 1.79-fold and 3.41-fold, respectively, with an increase in desipramine t 1/2 of 84 %.
The 3.29-fold and 3.41-fold increase in metoprolol and desipramine exposure, respectively, observed in these studies were less than the largest (approximately fivefold to sixfold) increases in AUC observed in studies of co-administration of metoprolol and desipramine with known strong CYP2D6 inhibitors: paroxetine (Laine et al. 2004; Alderman et al. 1997; Hemeryck et al. 2000; Nichols et al. 2009; Stout et al. 2011) and fluoxetine (Preskorn et al. 1994). This, together with the fact that the observed increases in metoprolol and desipramine AUC were between twofold and fivefold, and even the corresponding 90 % CIs were below the fivefold margin, demonstrates that the inhibitory effect of mirabegron towards CYP2D6 can be classified as moderate (European Medicines Agency 2010; US Food and Drug Administration 2012).
Because previous in vitro studies showed that mirabegron partly acts as an irreversible or quasi-irreversible, metabolism-dependent inhibitor of CYP2D6 (Takusagawa et al. 2012a), desipramine was administered for the third time 2 weeks after the last mirabegron dose. Although mirabegron concentrations were still quantifiable just prior to the third desipramine dose, albeit 200-fold lower than the steady-state mirabegron Cmax, C max and AUC for the third versus the first dose of desipramine were only about 1.13-fold increased and the 90 % CIs were within the range of 0.8–1.25. It can thus be concluded that at the latest after 2 weeks, the metabolic capacity was recovered. It is possible that the metabolic activity was recovered earlier, but this was not assessed in this study.
Due to the inhibition of the CYP2D6 metabolic pathway of metoprolol by mirabegron, a smaller amount could be converted to α-hydroxymetoprolol, hence its C max and AUC0-t decreased 2.6-fold and 2.2-fold, respectively. Graphically, the terminal phase of the α-hydroxymetoprolol plasma concentration–time curve with mirabegron co-treatment appeared not in parallel to the curve observed without mirabegron. It can be speculated that 160-mg mirabegron immediate release has a small effect on the clearance of α-hydroxymetoprolol, but this can also be due to the assay sensitivity.
Mean C max of the CYP2D6-mediated metabolite of desipramine, 2-hydroxydesipramine, was approximately 50 % lower in the presence of mirabegron and t max was prolonged with 1 h. The AUC0-∞ ratio of metabolite over parent decreased 2.5-fold. This indicates a slower conversion of desipramine into its 2-hydroxy metabolite, in line with CYP2D6 inhibition. It was accompanied by a 1.9-fold increase in 2-hydroxydesipramine half-life and a 1.3-fold increase in AUC0-∞. This AUC0-∞ increase has also been reported for other CYP2D6 inhibitors, e.g., paroxetine and deramciclane (Laine et al. 2004), and may indicate that a different route is involved under CYP2D6 inhibition. Fluvoxamine had no effect on desipramine clearance, which excludes CYP1A2, 2C9 and 2C19 (Spina et al. 1993). Interestingly, the inducers carbamazepine and phenobarbital induced the formation of 2-hydroxydesipramine (Spina et al. 1995, 1996) albeit CYP2D6 cannot be induced, suggesting the existence of an alternative elimination route. It is therefore possible that this alternative route is involved in the increased 2-hydroxydesipramine exposure. Another explanation would be that an alternative elimination route, not resulting in the formation of 2-hydroxydesipramine, is more strongly inhibited than CYP2D6. Under all conditions, the t 1/2 of 2-hydroxydesipramine was similar to that of desipramine, suggesting that its t 1/2 is determined by the rate of elimination of desipramine.
The exposure to desipramine, 2-hydroxydesipramine, and mirabegron was higher in female compared with male subjects in study 2. Although mirabegron exposure was higher in females than in males, the inhibitory effect of mirabegron on desipramine was not different. The exposure ratio of mirabegron to desipramine was similar for females and males. Correction for differences in body weight markedly reduced the magnitude of the sex differences in mirabegron oral clearance, with the remaining difference likely attributable to sex-related differences in P-glycoprotein-mediated efflux transport, in intestinal and/or hepatic first-pass metabolism (Eltink et al. 2012). For desipramine, BMI was a covariate in a population pharmacokinetic model (Gueorguieva et al. 2010), which may explain the sex difference.
The population in the drug–drug interaction studies comprised only CYP2D6 EMs. Because poor metabolizers have only minimal CYP2D6 activity, they are less likely to show the effects of CYP2D6 inhibition produced by compounds with inhibitory effects (Steiner and Spina 1987; Hamelin et al. 2000).
This study demonstrated a CYP2D6 inhibitory effect for both immediate-release and OCAS formulations of mirabegron. Immediate-release mirabegron would be expected to have a greater effect than mirabegron OCAS because the higher dose and increased drug input rate have greater potential to saturate CYP2D6 on hepatic first pass. However, the overall magnitude of drug interaction was approximately the same with the two formulations. This suggests that the exposure for both formulations was high enough to saturate CYP2D6. The mirabegron doses of 160 mg/day of the immediate-release formulation and 100 mg/day of the OCAS formulation were higher than the therapeutic dose of 50 mg OCAS to maximize the possibility of observing CYP2D6 inhibition.
All treatments were safe and well tolerated and mirabegron did not affect the cardiovascular responses to metoprolol or desipramine.
The results of the drug–drug interaction studies described here are consistent with inhibition of CYP2D6 enzymatic activity by mirabegron, resulting in an increase in exposure of both metoprolol and desipramine. The interaction was reversible, as after cessation of mirabegron treatment, activity of CYP2D6 returned to normal within 2 weeks. Data from the studies indicate that in vivo mirabegron can be classified as a moderate inhibitor of CYP2D6.
References
Aarnoutse RE, Kleinnijenhuis J, Koopmans PP, Touw D, Wieling J, Hekster YA, Burger DM (2005) Effect of low-dose ritonavir (100 mg twice daily) on the activity of cytochrome P450 2D6 in healthy volunteers. Clin Pharmacol Ther 78:664–674
Alderman J, Preskorn SH, Greenblatt DJ, Harrison W, Penenberg D, Allison J, Chung M (1997) Desipramine pharmacokinetics when coadministered with paroxetine or sertraline in extensive metabolizers. J Clin Psychopharmacol 17:284–291
Eltink C, Lee J, Schaddelee MP, Zhang W, Meijer J, van Marle S, Grunenberg N (2012) Single dose pharmacokinetics and absolute bioavailability of mirabegron, a selective and potent β3-adrenoceptor agonist for treatment of overactive bladder. Int J Clin Pharmacol Ther 50:838–850
European Medicines Agency (2010) Draft Guideline on the Investigation of Drug Interactions. http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/05/WC500090112.pdf. Accessed 28 January 2013
Evert B, Griese EU, Eichelbaum M (1994) Cloning and sequencing of a new non-functional CYP2D6 allele: deletion of T1795 in exon 3 generates a premature stop codon. Pharmacogenetics 4:271–274
Gueorguieva I, Jackson K, Wrighton SA, Sinha VP, Chien JY (2010) Desipramine, substrate for CYP2D6 activity: population pharmacokinetic model and design elements of drug–drug interaction trials. Br J Clin Pharmacol 70:523–536
Hamelin BA, Bouayad A, Méthot J, Jobin J, Desgagnés P, Poirier P, Allaire J (2000) Significant interaction between the nonprescription antihistamine diphenhydramine and the CYP2D6 substrate metoprolol in healthy men with high or low CYP2D6 activity. Clin Pharmacol Ther 67:466–477
Harris RZ, Salfi M, Posvar E, Hoelscher D, Padhi D (2007) Pharmacokinetics of desipramine HCl when administered with cinacalcet HCl. Eur J Clin Pharmacol 63:159–163
Heim M, Meyer UA (1990) Genotyping of poor metabolisers of debrisoquine by allele-specific PCR amplification. Lancet 336:529–532
Hemeryck A, Lefebvre RA, De Vriendt C, Belpaire FM (2000) Paroxetine affects metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Clin Pharmacol Ther 67:283–291
Johansson I, Lundqvist E, Dahl ML, Ingelman-Sundberg M (1996) PCR-based genotyping for duplicated and deleted CYP2D6 genes. Pharmacogenetics 6:351–355
Khullar V, Amarenco G, Angulo JC, Cambronero J, Hoye K, Milsom I, Radziszewski P, Rechberger T, Boerrigter P, Drogendijk T, Wooning M, Chapple C (2013) Efficacy and tolerability of mirabegron, a beta(3)-adrenoceptor agonist, in patients with overactive bladder: results from a Randomised European-Australian Phase 3 Trial. Eur Urol 63:283–295. doi:10.1016/j.eururo.2012.10.016
Laine K, De Bruyn S, Björklund H, Rouru J, Hänninen J, Scheinin H, Anttila M (2004) Effect of the novel anxiolytic drug deramciclane on cytochrome P(450) 2D6 activity as measured by desipramine pharmacokinetics. Eur J Clin Pharmacol 59:893–898
Lee J, Moy S, Meijer J, Krauwinkel W, Sawamoto T, Kerbusch V, Kowalski D, Roy M, Marion A, Takusagawa S, van Gelderen M, Keirns J. Role of Cytochrome P450 Isoenzymes 3A and 2D6 in the in Vivo Metabolism of Mirabegron, a β3-adrenoceptor agonist. Clin Drug Invest. 2013 (Accepted)
Lennard MS, Tucker GT, Silas JH, Woods HF (1986) Debrisoquine polymorphism and the metabolism and action of metoprolol, timolol, propranolol and atenolol. Xenobiotica 16:435–447
McGourty JC, Silas JH, Lennard MS, Tucker GT, Woods HF (1985) Metoprolol metabolism and debrisoquine oxidation polymorphism—population and family studies. Br J Clin Pharmacol 20:555–566
Nichols AI, Fatato P, Shenouda M, Paul J, Isler JA, Pedersen RD, Jiang Q, Ahmed S, Patroneva A (2009) The effects of desvenlafaxine and paroxetine on the pharmacokinetics of the cytochrome P450 2D6 substrate desipramine in healthy adults. J Clin Pharmacol 49:219–228
Nitti V, Auerbach S, Martin N, Calhoun A, Lee M, Herschorn S (2012) Results of a randomized phase III trial of mirabegron in patients with overactive bladder. J Urol. doi:10.1016/j.juro.2012.10.017 (Epub ahead of print)
Preskorn SH, Alderman J, Chung M, Harrison W, Messig M, Harris S (1994) Pharmacokinetics of desipramine coadministered with sertraline or fluoxetine. J Clin Psychopharmacol 14:90–98
Sallee FR, Pollock BG (1990) Clinical pharmacokinetics of imipramine and desipramine. Clin Pharmacokinet 18:346–364
Sawamoto T, Lee J, Alak A et al (2011) Phase I, open-label, drug interaction study to evaluate the effect of multiple doses of ketoconazole on single dose mirabegron (YM178) oral controlled absorption system (OCAS) in healthy adult subjects. Clin Pharmacol Ther 89(Suppl 1):S21
Schmid B, Bircher J, Preisig R, Küpfer A (1985) Polymorphic dextromethorphan metabolism: co-segregation of oxidative O-demethylation with debrisoquin hydroxylation. Clin Pharmacol Ther 38:618–624
Spina E, Pollicino AM, Avenoso A, Campo GM, Perucca E, Caputi AP (1993) Effect of fluvoxamine on the pharmacokinetics of imipramine and desipramine in healthy subjects. Ther Drug Monit 15:243–246
Spina E, Avenoso A, Campo GM, Caputi AP, Perucca E (1995) The effect of carbamazepine on the 2-hydroxylation of desipramine. Psychopharmacology 117:413–416
Spina E, Avenoso A, Campo GM, Caputi AP, Perucca E (1996) Phenobarbital induces the 2-hydroxylation of desipramine. Ther Drug Monit 18:60–64
Steijns LS, Van Der Weide J (1998) Ultrarapid drug metabolism: PCR-based detection of CYP2D6 gene duplication. Clin Chem 44:914–917
Steiner E, Spina E (1987) Differences in the inhibitory effect of cimetidine on desipramine metabolism between rapid and slow debrisoquin hydroxylators. Clin Pharmacol Ther 42:278–282
Stout SM, Nielsen J, Welage LS, Shea M, Brook R, Kerber K, Bleske BE (2011) Influence of metoprolol dosage release formulation on the pharmacokinetic drug interaction with paroxetine. J Clin Pharmacol 51:389–396
Takasu T, Ukai M, Sato S, Matsui T, Nagase I, Maruyama T, Sasamata M, Miyata K, Uchida H, Yamaguchi O (2007) Effect of (R)-2-(2-aminothiazol-4-yl)-4’-{2-[(2-hydroxy-2-phenylethyl)amino]ethyl} acetanilide (YM178), a novel selective beta3-adrenoceptor agonist, on bladder function. J Pharmacol Exp Ther 321:642–647. doi:10.1124/jpet.106.115840
Takusagawa S, Scheinkoenig J, Buckley D, Miyashita A, Iwatsubo T, Usui T (2012a) In vitro inhibition and induction of human cytochrome P450 enzymes by mirabegron, a potent and selective β3-adrenoceptor agonist. Xenobiotica 42:1187–1196
Takusagawa S, van Lier JJ, Suzuki K, Nagata M, Meijer J, Krauwinkel W, Schaddelee M, Sekiguchi M, Miyashita A, Iwatsubo T, van Gelderen M, Usui T (2012b) Absorption, metabolism and excretion of [(14)C]mirabegron (YM178), a potent and selective beta(3)-adrenoceptor agonist, after oral administration to healthy male volunteers. Drug Metab Dispos 40:815–824
Takusagawa S, Yajima K, Miyashita A, Iwatsubo T, Usui T (2012c) Identification of human CYP isoforms and esterases involved in the metabolism of mirabegron (YM178), a novel selective β3-adrenoceptor agonist. Xenobiotica 42(10):957–967
US Food and Drug Administration (2012) Draft guidance for industry on drug interaction studies—study design, data analysis, implications for dosing and labeling recommendations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed 28 January 2013
van Gelderen E, Li Q, Meijer J, Schaddelee MP, Takusagawa S, Sugawara T, de Koning P (2009) An exploratory comparison of the single dose pharmacokinetics of the β3-adrenoceptor agonist mirabegron in healthy CYP2D6 poor and extensive metabolizers. Clin Pharmacol Ther 85(Suppl 1):S88
Wieling J, Tamminga WJ, Sakiman EP, Oosterhuis B, Wemer J, Jonkman JH (2000) Evaluation of analytical and clinical performance of a dual-probe phenotyping method for CYP2D6 polymorphism and CYP3A4 activity screening. Ther Drug Monit 22:486–496
Yuen KH, Wong JW, Yap SP, Billa N (2001) Estimated coefficient of variation values for sample size planning in bioequivalence studies. Int J Clin Pharmacol Ther 39:37–40
Acknowledgments
The authors wish to thank Sandra Boom (Pharma-Plus) for her assistance in the preparation of this manuscript, funded by Astellas Pharma Europe.
Conflict of interest
Walter Krauwinkel, James Dickinson, Marloes Schaddelee, John Meijer, Reiner Tretter, and Marcel van Gelderen are employees of Astellas Pharma Europe BV. Dr. van de Wetering and Dr. Strabach have no disclosures to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Part of this research has been presented at the Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics in 2010.
Rights and permissions
About this article
Cite this article
Krauwinkel, W., Dickinson, J., Schaddelee, M. et al. The effect of mirabegron, a potent and selective β3-adrenoceptor agonist, on the pharmacokinetics of CYP2D6 substrates desipramine and metoprolol. Eur J Drug Metab Pharmacokinet 39, 43–52 (2014). https://doi.org/10.1007/s13318-013-0133-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-013-0133-1