
Abstract
Acute myeloid leukemia (AML) is a heterogeneous disorder among hematologic malignancies. Several genetic alterations occur in this disease, which cause proliferative progression, reducing differentiation and apoptosis in leukemic cells as well as increasing their survival. In the genetic study of AML, genetic translocations, gene overexpression, and mutations effective upon biology and pathogenesis of this disease have been recognized. Proto-oncogenes and tumor suppressor genes, which are important in normal development of myeloid cells, are involved in the regulation of cell cycle and apoptosis, undergo mutation in this type of leukemia, and are effective in prognosis of AML subtypes. This review deals with these genes, the assessment of which can be important in the diagnosis and prognosis of patients as well as therapeutic outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is developed due to somatically acquired genetic alterations in hematopoietic cells [1]. Genetic changes in AML result in proliferative progression as well as reducing leukemic cell differentiation and apoptosis [2]. Excessive proliferation of stem or progenitor cells causes replacement of normal erythroid, myeloid, and megakaryocytic precursors with malignant cells, which gives rise to hematopoietic deficiency (i.e., granulocytopenia, thrombocytopenia, or anemia) in the bone marrow (BM) and peripheral blood [3, 4]. Approximately 20,000 patients were diagnosed with AML with over 10,000 death cases of AML patients in the USA in 2015 [5]. AML is more frequently seen in the elderly. The incidence of AML in the USA is 3.5 cases per 100,000 people, being higher in patients >65 years compared with younger patients (15.9 vs 1.7 cases, respectively) [6].
Mutations in this disease can be divided into two categories: (1) mutation in the genes involved in cell proliferation and survival such as mutations of FLT3, oncogenic Ras, PTPN11, and TEL/PDGFbR gene fusions and (2) mutations affecting differentiation and apoptosis, such as AML/ETO and PML/RARa fusions, MLL rearrangements, mutations in CEBPA, CBF, HOX family members, CBP/P300, and coactivators of TIF1 [7].
Proto-oncogenes and tumor suppressor genes encode the proteins involved in the regulation of cell surface receptors for cytokines, growth factors, signal transduction molecules, transcription factors, as well as epigenetic regulators and regulators of cell cycle and apoptosis [3]. Typically, most of these genes are involved in normal development of myeloid cell, so any disruption in their expression or loss of their normal function leads to leukemogenesis [8]. For example, Flt3 (the receptor tyrosine kinase) is expressed in early hematopoietic progenitor cells and plays important roles in proliferation and survival [9]. This receptor shows significant functions associated with c-kit and stem cell factor receptor. In cooperation with lineage-specific cytokines, Flt3 activation increases colony-forming capacity of all hematopoietic lineages [10]. Furthermore, several gene mutations were found to cause epigenetic changes and to deregulate gene expression in AML, such as mutations of the TET2 gene as well as IDH1 and DNMT3A mutations [11].
Studies show that approximately 35 % of AML patients have several translocations causing oncofusion proteins. Transcription of these proteins could target mechanisms such as transcription, epigenetics, cell structure, and nuclear receptors as well as causing uncontrolled proliferation of progenitor cells [12]. There are four important translocations in AML with a frequency of 3–10 %, including PML-RARa, AML1-ETO, CBFb-MYH11, and MLL-fusions as well as other oncofusion proteins with a lower incidence [13]. For example, t(15; 17) together with fusion of RARA (retinoic acid receptor alpha) gene with a previously unknown gene designated as PML (promyelocytic leukemia) encodes an oncofusion protein effective in the regulation of apoptosis and prevention of cell differentiation [14]. Another common translocation in AML is t(8;21) (q22;q22), which gives rise to acute myeloid gene 1 (AML1) and ETO (eight twenty-one) [15]. The AML1 gene encodes a critical transcription factor that regulates a variety of genes involved in proliferation and differentiation of many cell types, including those within the hematopoietic system [16]. On the other hand, ETO is a protein-harboring transcriptional repressor activities. Consequently, AML1-ETO functions as a transcriptional repressor [17].
Various cytogenetic aberrations occur in AML, which are very important for the prognosis of patients and predict the possibility of response to treatment or relapse in patients [11]. These are divided to three groups: (1) favorable risk: t(8; 21) (q22; q22), inv [16] (p13q22)/t(16; 16) (p13; q22), or t(15; 17) (q22; q21); (2) intermediate risk: normal cytogenetics, +8, t(9; 11); and (3) unfavorable risk: −7, inv [3] (q21q26)/t(3; 3) (q21; q26), balanced translocations involving 11q23 other than t(9; 11) (p22; q23) or complex karyotype [18, 19]. Delaunay et al. studied 110 patients with inv [16]/t(16; 16) AML with a complete remission (CR) rate of 93 % [20]. Clozel et al. have introduced the association of this type of AML with an overall good prognosis; however, relapse still occurs in 30–35 % of patients and with a higher frequency in older patients [21]. In addition, adverse prognostic factors include increasing age, a poor performance before treatment, unfavorable cytogenetic abnormalities, and a high white blood cell count. Also, therapy-related AML or AML with a myelodysplastic or myeloproliferative syndrome history is more resistant to usual treatments than de novo AML [11].
Molecular and cytological studies show that genetic mutations in AML patients are the most important prognostic factors for predicting clinical outcome of patients [1]. Therefore, these changes may be used as diagnostic and prognostic markers. In this article, we have attempted to introduce the prognostic role of oncogenes and tumor suppressor genes in AML cells.
Oncogenes in AML
Mutations in proto-oncogenes lead to excessive proliferation of myeloid cells in leukemia [22]. Some oncogenes encode hematopoietic growth factors or growth factor receptors like FLT3, and some others regulate cell proliferation or differentiation (e.g., RAS). Mutation, translocation, and amplification in these important cell processes contribute to leukemogenesis [23, 24]. Abnormalities in cellular oncogenes have been reported in leukemia, and the most important reported AML oncogenes are herewith evaluated (Table 1). Given the importance of these genes in the diagnosis of disease, they can contribute to monitoring of disease and determination of prognosis as MRD markers [25].
Mutation in FMS-like tyrosine kinase 3 (FLT3) receptor gene, which encodes a membrane protein of type III platelet-derived growth factor (PDGF) family, is an important mutation in AML. FLT3 binding to its ligand and its subsequent activation induces cell proliferation and survival [18, 26]. In addition, this protein plays a role in hematopoiesis and malignant transformation of primitive hematopoietic cells [27, 28]. Mutation in this gene occurs in about 30 % of AML cases in two forms: internal tandem duplication (ITD) and tyrosine kinase domain (TKD). It occurs as a result of duplication and insertion of juxta membrane domain sequence with ≈20–30 % incidence in AML patients and following missense point mutation within the activation loop of the second TKD with lower incidence, respectively [29–31]. The high prevalence of FLT3-ITDs in AML patients raises it as a common marker detectable by PCR [32]. Studies have shown that FLT3-ITD detection is associated with increased BM blasts and white blood cell (WBC) count in peripheral blood; therefore, the death rate and relapse risk is increased, which is a sign of poor outcome in normal karyotype AML (NK-AML) [33]. If this mutation is detected in AML cases, inhibition of its downstream pathways, including AKT, BAD, BCL2, and STAT5[18] can help improve the specific treatment process via suppression of this gene, which may be used to evaluate the prognosis of patients, especially in cases with normal karyotype.
AML1 (CBFA2/PEBP2αB/RUNX1) is another gene undergoing mutation in acute leukemias (Table 1) [34]. The encoded AML1 protein combined with a common heterodimeric binding cofactor (CBFβ) is attached to a specific DNA sequence TGT/cGGT and regulates the expression of genes effective upon hematopoiesis, including granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-3 (IL-3), and colony-stimulating factor 1 (CSF1) receptor [35]. This protein is an important regulator of progenitor cell fate and marks the proliferation, differentiation, and apoptosis pathways in the cell [36]. This gene is involved in both normal and malignant hematopoiesis. In leukemia, this subunit undergoes t(8; 21), t(3; 21), and t(16; 21) translocation. AML1/ETO t(8; 21) (q22; q22) fusion is observed in nearly 10–15 % of AML cases. This fusion inhibits the proliferation and differentiation of hematopoietic cells and increases self-renewal of hematopoietic stem cells [37]. Assessment of AML1 mutations in de novo AML is associated with male sex, older age, immature FAB subtypes, and trisomy 8, which are associated with poor prognosis during treatment [38].
SALL4 is another oncogene involved in AML, which belongs to the four-member family of SALL1 to SALL4. SALL protein is among C2H2 zinc finger transcription factors [39]. This gene family is involved in normal hematopoiesis and development of cells [40]. SALL4 isoform has been reported in many hematologic malignancies, including AML, precursor B cell lymphoblastic leukemia/lymphoma, and myelodysplastic syndrome (MDS) [41, 42]. SALL4 directly regulates the expression of apoptotic genes such as TP53, BCL2, TNF, and PTEN, indicating its role in leukemogenesis [43]. Investigation of NB4 leukemic cell line (M3) indicated that the apoptosis pathway was induced in cells with a low expression level of this gene and caused cell cycle arrest [44]. Reduction of SALL4 level causes overexpression of SALL4 downstream target protein of Bmi-1 and maintains the apoptosis capacity of the cell. These epigenetic alterations in the methylation of SALL4 gene promoter are able to induce apoptosis in the cells [43, 45]. SALL4 is differently expressed in various subgroups of AML, causing acquisition and maintenance of blastic traits such as self-renewal and/or lack of differentiation in leukemic stem cells (LSCs), which is associated with older age and increased WBC count of patients [46, 47]. Increased expression of SALL4 in AML patients leads to a worse prognosis via induction of drug resistance [48]. Therefore, according to the regulatory role of this gene in leukemic cell survival, downregulation of SALL4 can significantly induce cell apoptosis, and the importance of evaluating the expression level of SALL4 as a new method to predict prognosis and response to treatment in patients is thus highlighted.
Given the foregoing, prognostic significance of oncogenes in AML is understood, which can be used as markers for monitoring diagnosis and monitoring of patients.
Tumor suppressor genes in AML
Tumor suppressor genes encode proteins with inhibitory roles in the cell cycle. Loss-of-function mutations in these genes cause uncontrolled proliferation of cells and promotion of malignancy [49]. Tumor suppressor genes are generally known as negative regulators of cell growth effective upon invasive and metastatic ability [50]. In this section, a number of important tumor suppressor genes mutated in AML will be discussed.
WT1 transcription factor is important in cell growth and development with different expression levels in various stages of cell development. For example, it is expressed during embryonic development in the urogenital system and is expressed in adult urogenital system, central nervous system, and hematopoietic tissues like BM and lymph nodes [51]. WT1 has a low expression level in CD34+ cells in normal human BM and acts as a tumor suppressor gene [52]. When a deletion occurs in the second zinc finger of this protein, its expression is increased in early human BM cells, which results in growth arrest and reduced colony formation [53, 54]. WT1 is expressed in several leukemia types and can be evaluated to detect residual disease. WT1 mutations have been reported with different frequencies in heterozygous, homozygous, and compound heterozygous forms in several adult and childhood AML cases [55]. These mutations have been detected in cytogenetically normal AML or in combination with other mutations such as FLT3 [56, 57]. Mutation in WT1 may even be detected during relapse in patients who do not show it upon diagnosis [58]. Higher levels of this protein are associated with decreased attainment of remission, poor disease-free survival, and/or poor overall survival [59] (Table 2).
The promyelocytic leukemia (PML) gene (15q22) is a tumor suppressor gene present in normal cells as nuclear structures called PML-nuclear bodies (PML-NBs) [60][50]. Cell cycle regulation, viral infections, growth inhibition, tumor suppression, apoptosis, and transcriptional regulation are among the intracellular functions of these PML-NBs, which are involved in the acetylation of P53 tumor suppressor and regulation of the oncogenic function of Ras [61, 62]. PML-4 (isoform IV) has a more prominent tumor suppressor role and can efficiently inhibit the transcription of anti-apoptotic proteins such as survivin as well as apoptosis signaling by binding to regulators of apoptotic genes like histone deacetylases (HDAC) [63–65]. This gene is fused with retinoic acid receptor alpha (RARα) gene in acute promyelocytic leukemia (APL) sub-group and encodes the t(15; 17) oncofusion protein [63], which is detected in 97 % of APL patients. The protein resulting from this translocation inhibits differentiation of myeloid hematopoietic cells via suppression of PU.1 [66, 67]. In general, the presence of this fusion protein causes good prognosis in the patient. PML-RARA fusion can be used for both diagnosis and detection of minimal residual disease [68]. Measurement of PML/RARα fusion gene is important to predict relapse even in the absence of t(15; 17) by karyotyping and fluorescent in situ hybridization (Table 2).
Tet oncogene family member 2 (TET2) is mutated in a variety of hematologic disorders such as MDS, myeloproliferative neoplasms (MPN), chronic myeloid leukemia (CML), and AML, which has been reported secondarily in AML with a history of MDS/MPN [69, 70]. Mutations in this gene are usually observed in the form of deletion or uniparental disomy [71], increasing the self-renewal capacity of LSCs and causing defective hematopoiesis, monocytosis, and extramedullary hematopoiesis [72]. TET2 mutations are associated with older patients, higher WBC and blast counts, low platelet count, normal karyotype, intermediate-risk cytogenetics, as well as mutation in NPM1 and ASXL1 but exclusively with IDH mutation. Studies show that mutation in TET2 is developed due to IDH mutation as an epigenetic factor in AML [73–75]. TET2 gene has been associated with poor prognosis in AML patients but is considered as a good prognostic factor in MDS patients with trisomy 8 [76]. The difference in the relationship between this molecule with other molecules mutated in AML and various diseases indicates the prognostic value of this marker. TET1 is another family member of TET reported in t(10; 11) (q22; q23) translocation in some AML cases, which is developed due to fusion between TET1 and MLL in 11q23 position and belongs to acute myelomonocytic leukemia (FAB. M5) subgroup [77]. Therefore, according to the above, diagnostic and prognostic importance of tumor suppressor genes in AML patients is understood, which can pave the way for faster and more specific detection in each of the AML subtypes (Table 2).
Discussion and future perspective
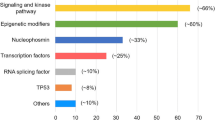
AML is developed due to accumulation of abnormal blast cells in the BM, increased proliferation, and self-renewal which leads to hematopoietic insufficiency [4, 78]. So far, over 200 disorders have been reported in AML patients, which are cytogenetic findings important in the prognosis of patients (Fig. 1) [4]. In general, the factors affecting prognosis include age, initial leukocyte count, karyotype, immune phenotype, and response to remission-induction therapy. If the patient is older, has a higher WBC count, and does not respond to treatment, an adverse prognosis is expected for them [79, 80]. Mutations in AML patients are important for the diagnosis and prognosis of AML patients and are specifically introduced for each AML subtype. These molecular markers can also be used to monitor and evaluate patient’s CR rate, relapse risk (RR), and overall survival (OS) [81].

Genetic aberrations in AML. In this type of leukemia, genetic alterations occur in three forms of fusion genes, mutations, and overexpression. Oncofusions such as PML-RARA are fusion results of two different genes. In some of the identified genes like FLT3, point mutations alter the gene function. The expression of the third category of genes is increased, which mostly contribute to epigenetic mechanisms in AML stem cells.
Given the role of oncogenes in the proliferation, differentiation, apoptosis, and survival of hematopoietic progenitors, they can give rise to several cancers such as leukemia if they are subject to mutation. Oncogene mutation with varied frequency is observed in AML subgroups. Considering Table 1, prognosis is poor in case of the presence of these genes even in favorable risk AML subgroup, including mutations in KIT [82].
Mutation of tumor suppressor genes in addition to factors such as patient’s age, translocations, and the involved cell line can be important markers for assessing the prognosis of patients. As can be seen in Table 2, mutation in these genes causes a poor prognosis; however, if mutation is detected in C/EBPα transcription factor, which is common in favorable risk disease group, a better prognosis is expected even in older patients [83]. Moreover, some of these genes are associated with patients’ age, for example, mutation in TET2 and IDH1 has not been reported in childhood AML [75, 84]. Thus, general understanding of molecular mechanisms responsible for leukemia would help design more specific diagnostic methods, better monitoring of disease prognosis, and treatment protocol.
In the past, cytogenetic factors were used as the most important prognostic factors to assess response to treatment and survival of patients. Today, a large number of molecular markers have been identified for molecular risk classification of AML. As a result, many reported results from studies need to be assessed and confirmed with more samples to introduce the most reliable available markers.
References
Grimwade D. The changing paradigm of prognostic factors in acute myeloid leukaemia. Best Pract Res Clin Haematol. 2012;25(4):419–25.
Dash A, Gilliland DG. Molecular genetics of acute myeloid leukaemia. Best Pract Res Clin Haematol. 2001;14(1):49–64.
Lo-Coco F, Breccia M, Noguera N, Miller Jr WH. Diagnostic value of detecting fusion proteins derived from chromosome translocations in acute leukaemia. Best Pract Res Clin Haematol. 2003;16(4):653–70.
De Jonge H, Huls G, De Bont E. Gene expression profiling in acute myeloid leukaemia. Neth J Med. 2011;69(4):167–76.
Ramos NR, Mo CC, Karp JE, Hourigan CS. Current approaches in the treatment of relapsed and refractory acute myeloid leukemia. J Clin Med. 2015;4(4):665–95.
Villela L, Bolaños-Meade J. Acute myeloid leukaemia: optimal management and recent developments. Drugs. 2011;71(12):1537–50.
Rubnitz JE, Gibson B, Smith FO. Acute myeloid leukemia. Hematology/oncology clinics of North America. 2010;24(1):35–63.
Willman CL, Whittaker MH. The molecular biology of acute myeloid leukemia. Proto-oncogene expression and function in normal and neoplastic myeloid cells. Clin Lab Med. 1990;10(4):769–96.
Lyman SD, Jacobsen SEW. c-kit ligand and Flt3 ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91(4):1101–34.
Mizuki M, Schwäble J, Steur C, Choudhary C, Agrawal S, Sargin B, et al. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood. 2003;101(8):3164–73.
Bullinger L. New avenues for genetics guided therapeutic approaches in AML. Acta Haematol Pol. 2014;45(4):322–9.
Martens JH, Stunnenberg HG. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett. 2010;584(12):2662–9.
Redner RL, Wang J, Liu JM. Chromatin remodeling and leukemia: new therapeutic paradigms. Blood. 1999;94(2):417–28.
Brown N, Ramalho M, Pedersen EW, Moravcsik E, Solomon E, Grimwade D. PML nuclear bodies in the pathogenesis of acute promyelocytic leukemia: active players or innocent bystanders? Front Biosci (Landmark edition). 2008;14:1684–707.
Michaud J, Scott HS, Escher R. AML1 interconnected pathways of leukemogenesis. Cancer Investig. 2003;21(1):105–36.
Lo Coco F, Pisegna S, Diverio D. The AML1 gene: a transcription factor involved in the pathogenesis of myeloid and lymphoid leukemias. Haematologica. 1997;82(3):364–70.
Fenske TS, Pengue G, Graubert TA. Dominant negative effects of the AML1/ETO fusion oncoprotein. Cell Cycle. 2005;4(1):33–6.
Stone RM. Prognostic factors in AML in relation to (ab) normal karyotype. Best Pract Res Clin Haematol. 2009;22(4):523–8.
Blum W, Marcucci G. New approaches in acute myeloid leukemia. Best Pract Res Clin Haematol. 2008;21(1):29–41.
Delaunay J, Vey N, Leblanc T, Fenaux P, Rigal-Huguet F, Witz F, et al. Prognosis of inv(16)/t(16;16) acute myeloid leukemia (AML): a survey of 110 cases from the French AML Intergroup. Blood. 2003;102(2):462–9.
Clozel T, Renneville A, Venot M, Gardin C, Kelaidi C, Leroux G, et al. Slow relapse in acute myeloid leukemia with inv(16) or t(16;16). Haematologica. 2009;94(10):1466–7.
Hartl M, Bister K. Oncogenes. In: Hughes SM, editor. Brenner’s encyclopedia of genetics. 2nd ed. San Diego: Academic Press; 2013. p. 164–6.
Carow CE, Levenstein M, Kaufmann SH, Chen J, Amin S, Rockwell P, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood. 1996;87(3):1089–96.
Zhang H, Alberich-Jorda M, Amabile G, Yang H, Staber Philipp B, Di Ruscio A, et al. Sox4 is a key oncogenic target in C/EBPα mutant acute myeloid leukemia. Cancer Cell. 2013;24(5):575–88.
Butturini A, Gale RP. Oncogenes and leukemia. Leukemia. 1990;4(2):138–60.
Shih L, Huang C, Wang P, Wu J, Lin T, Dunn P, et al. Acquisition of FLT3 or N-ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia. 2004;18(3):466–75.
Shih L-Y, Huang C-F, Wu J-H, Lin T-L, Dunn P, Wang P-N, et al. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100(7):2387–92.
Whitman SP, Ruppert AS, Radmacher MD, Mrózek K, Paschka P, Langer C, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008;111(3):1552–9.
Fröhling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23(26):6285–95.
Breitenbuecher F, Schnittger S, Grundler R, Markova B, Carius B, Brecht A, et al. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood. 2009;113(17):4074–7.
Scholl S, Loncarevic IF, Krause C, Kunert C, Clement JH, Höffken K. Minimal residual disease based on patient specific Flt3-ITD and -ITT mutations in acute myeloid leukemia. Leuk Res. 2005;29(7):849–53.
Stirewalt DL, Willman CL, Radich JP. Quantitative, real-time polymerase chain reactions for FLT3 internal tandem duplications are highly sensitive and specific. Leuk Res. 2001;25(12):1085–8.
Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Am Soc Hematol Educ Program Book. 2013;2013(1):220–6.
Nguyen LA, Pandolfi PP, Aikawa Y, Tagata Y, Ohki M, Kitabayashi I. Physical and functional link of the leukemia-associated factors AML1 and PML. Blood. 2005;105(1):292–300.
Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X, et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87(4):697–708.
Cameron ER, Neil JC. The Runx genes: lineage-specific oncogenes and tumor suppressors. Oncogene. 2004;23(24):4308–14.
Higuchi M, O’Brien D, Kumaravelu P, Lenny N, Yeoh E-J, Downing JR. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell. 2002;1(1):63–74.
Tang J-L, Hou H-A, Chen C-Y, Liu C-Y, Chou W-C, Tseng M-H. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114(26):5352–61.
Al-Baradie R, Yamada K, Hilaire CS, Chan W-M, Andrews C, McIntosh N, et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet. 2002;71(5):1195–9.
Wang F, Guo Y, Chen Q, Yang Z, Ning N, Zhang Y, et al. Stem cell factor SALL4, a potential prognostic marker for myelodysplastic syndromes. J Hematol Oncol. 2013;6:73.
Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270.
Merup M, Lazarevic V, Nahi H, Andreasson B, Malm C, Nilsson L, et al. Different outcome of allogeneic transplantation in myelofibrosis using conventional or reduced‐intensity conditioning regimens. Br J Haematol. 2006;135(3):367–73.
Yang J, Chai L, Gao C, Fowles TC, Alipio Z, Dang H, et al. SALL4 is a key regulator of survival and apoptosis in human leukemic cells. Blood. 2008;112(3):805–13.
Shuai X, Zhou D, Shen T, Wu Y, Zhang J, Wang X, et al. Overexpression of the novel oncogene SALL4 and activation of the Wnt/β-catenin pathway in myelodysplastic syndromes. Cancer Genet Cytogenet. 2009;194(2):119–24.
Yang J, Chai L, Liu F, Fink LM, Lin P, Silberstein LE, et al. Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc Natl Acad Sci. 2007;104(25):10494–9.
Chen Q, Qian J, Lin J, Yang J, Li Y, Wang C, et al. Expression of SALL4 gene in patients with acute and chronic myeloid leukemia. Zhongguo shi yan xue ye xue za zhi/Zhongguo bing li sheng li xue hui. Journal of Experimental Hematology/Chinese Association of Pathophysiology. 2013;21(2):315–9.
Ma J-c, Qian J, Lin J, Qian W, Yang J, Wang C-z, et al. Aberrant hypomethylation of SALL4 gene is associated with intermediate and poor karyotypes in acute myeloid leukemia. Clin Biochem. 2013;46(4):304–7.
Jeong H-W, Cui W, Yang Y, Lu J, He J, Li A, et al. SALL4, a stem cell factor, affects the side population by regulation of the ATP-binding cassette drug transport genes. PLoS ONE. 2011;6(4):e18372.
Hinds PW, Weinberg RA. Tumor suppressor genes. Curr Opinion Gen Dev. 1994;4(1):135–41.
Osborne C, Wilson P, Tripathy D. Oncogenes and tumor suppressor genes in breast cancer: potential diagnostic and therapeutic applications. Oncologist. 2004;9(4):361–77.
Menke AL, Van der Eb A, Jochemsen A. The Wilms’ tumor 1 gene: oncogene or tumor suppressor gene? Int Rev Cytol. 1998;181:151–212.
Hosen N, Sonoda Y, Oji Y, Kimura T, Minamiguchi H, Tamaki H, et al. Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br J Haematol. 2002;116(2):409–20.
Owen C, Fitzgibbon J, Paschka P. The clinical relevance of Wilms Tumour 1 (WT1) gene mutations in acute leukaemia. Hematol Oncol. 2010;28(1):13–9.
Svensson E, Eriksson H, Gekas C, Olofsson T, Richter J, Gullberg U. DNA-binding dependent and independent functions of WT1 protein during human hematopoiesis. Exp Cell Res. 2005;308(1):211–21.
Huff V. Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer. 2011;11(2):111–21.
Summers K, Stevens J, Kakkas I, Smith M, Smith L, Macdougall F, et al. Wilms’ tumour 1 mutations are associated with FLT3-ITD and failure of standard induction chemotherapy in patients with normal karyotype AML. Leukemia. 2007;21(3):550–1.
Paschka P, Marcucci G, Ruppert AS, Whitman SP, Mrózek K, Maharry K, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2008;26(28):4595–602.
Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, Balgobind BV, Arentsen-Peters ST, Alders M, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood. 2009;113(23):5951–60.
Ellisen LW, Carlesso N, Cheng T, Scadden DT, Haber DA. The Wilms tumor suppressor WT1 directs stage‐specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 2001;20(8):1897–909.
Martin-Martin N, Sutherland JD, Carracedo A. PML: not all about tumor suppression. Front Oncol. 2013;3:200.
Sternsdorf T, Grötzinger T, Jensen K, Will H. Nuclear dots: actors on many stages. Immunobiology. 1997;198(1):307–31.
Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito SI, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406(6792):207–10.
Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108(2):165–70.
Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene. 2001;20(49):7223–33.
Gamell C, Jan Paul P, Haupt Y, Haupt S. PML tumour suppression and beyond: therapeutic implications. FEBS Lett. 2014;588(16):2653–62.
Bernardi R, Pandolfi PP. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene. 2003;22(56):9048–57.
Mueller BU, Pabst T, Fos J, Petkovic V, Fey MF, Asou N, et al. ATRA resolves the differentiation block in t(15;17) acute myeloid leukemia by restoring PU.1 expression. Blood. 2006;107(8):3330–8.
Meloni G, Diverio D, Vignetti M, Avvisati G, Capria S, Petti MC, et al. Autologous bone marrow transplantation for acute promyelocytic leukemia in second remission: prognostic relevance of pretransplant minimal residual disease assessment by reverse-transcription polymerase chain reaction of the PML/RARα fusion gene. Blood. 1997;90(3):1321–5.
Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–10.
Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–7.
Delhommeau F, Dupont S, Valle VD, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24.
Chou W-C, Chou S-C, Liu C-Y, Chen C-Y, Hou H-A, Kuo Y-Y, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118(14):3803–10.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67.
Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26(5):934–42.
Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood. 2009;114(15):3285–91.
Lorsbach R, Moore J, Mathew S, Raimondi S, Mukatira S, Downing J. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t (10; 11)(q22; q23). Leukemia. 2003;17(3):637–41.
Estey EH. Acute myeloid leukemia: 2013 update on risk‐stratification and management. Am J Hematol. 2013;88(4):317–27.
Zheng J, Wang X, Hu Y, Yang J, Liu J, He Y, et al. A correlation study of immunophenotypic, cytogenetic, and clinical features of 180 AML patients in China. Cytometry B Clin Cytom. 2008;74(1):25–9.
Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93(9):3074–80.
Gaidzik V, Döhner K. Prognostic implications of gene mutations in acute myeloid leukemia with normal cytogenetics. Semin Oncol. 2008;35(4):346–55. doi: 10.1053/j.seminoncol.2008.04.005.
Paschka P, Marcucci G, Ruppert AS, Mrózek K, Chen H, Kittles RA, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv (16) and t (8; 21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24(24):3904–11.
Preudhomme C, Sagot C, Boissel N, Cayuela J-M, Tigaud I, de Botton S, et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood. 2002;100(8):2717–23.
Liang D-C, Liu H-C, Yang C-P, Jaing T-H, Hung I-J, Yeh T-C, et al. Cooperating gene mutations in childhood acute myeloid leukemia with special reference on mutations of ASXL1, TET2, IDH1, IDH2, and DNMT3A. Blood. 2013;121(15):2988–95.
Gao C, Dimitrov T, Yong KJ, Tatetsu H, Jeong H-W, Luo HR, et al. Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood. 2013;121(8):1413–21.
Chong PS, Zhou J, Cheong L-L, Liu S-C, Qian J, Guo T, et al. LEO1 is regulated by PRL-3 and mediates its oncogenic properties in acute myelogenous leukemia. Cancer Res. 2014;74(11):3043–53.
Park JE, Yuen HF, Zhou JB, Al‐aidaroos AQO, Guo K, Valk PJ, et al. Oncogenic roles of PRL-3 in FLT3-ITD induced acute myeloid leukaemia. EMBO Mol Med. 2013;5(9):1351–66.
Qu S, Liu B, Guo X, Shi H, Zhou M, Li L, et al. Independent oncogenic and therapeutic significance of phosphatase PRL-3 in FLT3-ITD–negative acute myeloid leukemia. Cancer. 2014;120(14):2130–41.
Gari M, Goodeve A, Wilson G, Winship P, Langabeer S, Linch D, et al. c-kit proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol. 1999;105(4):894–900.
Liu D, Jiang H, Qin Y-Z, Xu L-P, Jiang Q, Zhang X-H, et al. KIT mutation versus MRD, which is more important to predict relapse of acute myeloid leukemia with t (8; 21)? Blood. 2013;122(21):1309–9.
Naoe T, Kiyoi H. Normal and oncogenic FLT3. Cell Mol Life Sci. 2004;61(23):2932–8.
Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100(1):59–66.
Kainz B, Heintel D, Marculescu R, Schwarzinger I, Sperr W, Le T, et al. Variable prognostic value of FLT3 internal tandem duplications in patients with de novo AML and a normal karyotype, t (15; 17), t (8; 21) or inv (16). Hematol J. 2002;3(6):283–9.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–9.
Chan IT, Gilliland DG. Oncogenic K-ras in mouse models of myeloproliferative disease and acute myeloid leukemia. Cell Cycle. 2004;3(5):534–5.
Neubauer A, Dodge R, George S, Davey F, Silver R, Schiffer C, et al. Prognostic importance of mutations in the ras proto-oncogenes in de novo acute myeloid leukemia. Blood. 1994;83(6):1603–11.
Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97(11):3589–95.
Lutterbach B, Hiebert S. Role of the transcription factor AML-1 in acute leukemia and hematopoietic differentiation. Gene. 2000;245(2):223–35.
Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–30.
Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123(9):3876.
Mendler JH, Maharry K, Radmacher MD, Mrózek K, Becker H, Metzeler KH, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and microRNA expression signatures. J Clin Oncol. 2012;30(25):3109–18.
Schessl C, Rawat VPS, Cusan M, Deshpande A, Kohl TM, Rosten PM, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest. 2005;115(8):2159–68.
Wieser R. The oncogene and developmental regulator EVI1: Expression, biochemical properties, and biological functions. Gene. 2007;396(2):346–57.
Nucifora G. The EVI1 gene in myeloid leukemia. Leukemia. 1997;11(12):2022–31.
Lennon PA, Abruzzo LV, Medeiros LJ, Cromwell C, Zhang X, Yin CC, et al. Aberrant EVI1 expression in acute myeloid leukemias associated with the t (3; 8)(q26; q24). Cancer Genet Cytogenet. 2007;177(1):37–42.
Gröschel S, Lugthart S, Schlenk RF, Valk PJM, Eiwen K, Goudswaard C, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol. 2010;28(12):2101–7.
Shearer BM, Knudson RA, Flynn HC, Ketterling RP. Development of a D-FISH method to detect DEK/CAN fusion resulting from t(6;9)(p23;q34) in patients with acute myelogenous leukemia. Leukemia. 2005;19(1):126–31.
Logan GE, Mor-Vaknin N, Braunschweig T, Jost E, Schmidt PV, Markovitz DM, et al. DEK oncogene expression during normal hematopoiesis and in acute myeloid leukemia (AML). Blood Cell Mol Dis. 2015;54(1):123–31.
Sandahl JD, Coenen EA, Forestier E, Harbott J, Johansson B, Kerndrup G, et al. t (6; 9)(p22; q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international study of 62 patients. Haematologica. 2014;99(5):865–72.
Heuser M, Beutel G, Krauter J, Döhner K, von Neuhoff N, Schlegelberger B, et al. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood. 2006;108(12):3898–905.
Grosveld GC. MN1, a novel player in human AML. Blood Cells Mol Dis. 2007;39(3):336–9.
Liu T, Jankovic D, Brault L, Ehret S, Baty F, Stavropoulou V, et al. Functional characterization of high levels of meningioma 1 as collaborating oncogene in acute leukemia. Leukemia. 2010;24(3):601–12.
Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7(4):351–62.
Siu Y-T, Jin D-Y. CREB—a real culprit in oncogenesis. FEBS J. 2007;274(13):3224–32.
Kinjo K, Sandoval S, Sakamoto KM, Shankar DB. The role of CREB as a proto-oncogene in hematopoiesis. Cell Cycle. 2005;4(9):1134–5.
Ho PA, Alonzo TA, Kopecky KJ, Miller KL, Kuhn J, Zeng R, et al. Molecular alterations of the IDH1 gene in AML: a Children’s Oncology Group and Southwest Oncology Group study. Leukemia. 2010;24(5):909–13.
Chou W-C, Huang H-H, Hou H-A, Chen C-Y, Tang J-L, Yao M, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116(20):4086–94.
Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118(26):6920–9.
Metzeler KH, Maharry K, Radmacher MD, Mrózek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29(10):1373–81.
Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci. 2014;105(9):1093–9.
Hou H-A, Kuo Y-Y, Liu C-Y, Chou W-C, Lee MC, Chen C-Y, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119(2):559–68.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33.
Shivarov V, Gueorguieva R, Stoimenov A, Tiu R. DNMT3A mutation is a poor prognosis biomarker in AML: results of a meta-analysis of 4500 AML patients. Leuk Res. 2013;37(11):1445–50.
King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms’ tumor gene WT1 in leukemias. Blood. 1996;87(6):2171–9.
Yang L, Han Y, Saiz FS, Minden M. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21(5):868–76.
Trka J, Kalinova M, Hrusak O, Zuna J, Krejci O, Madzo J, et al. Real-time quantitative PCR detection of WT1 gene expression in children with AML: prognostic significance, correlation with disease status and residual disease detection by flow cytometry. Leukemia. 2002;16(7):1381–9.
Bally C, Adès L, Renneville A, Sebert M, Eclache V, Preudhomme C, et al. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk Res. 2014;38(7):751–5.
Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19(5):1405–13.
Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106(9):3150–9.
Zhao Z, Zuber J, Diaz-Flores E, Lintault L, Kogan SC, Shannon K, et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes Dev. 2010;24(13):1389–402.
Puccetti E, Ruthardt M. Acute promyelocytic leukemia: PML//RAR[alpha] and the leukemic stem cell. Leukemia. 2004;18(7):1169–75.
Wen X-M, Lin J, Yang J, Yao D-M, Deng Z-Q, Tang C-Y, et al. Double CEBPA mutations are prognostically favorable in non-M3 acute myeloid leukemia patients with wild-type NPM1 and FLT3-ITD. Int J Clin Exp Pathol. 2014;7(10):6832.
Ho PA, Alonzo TA, Gerbing RB, Pollard J, Stirewalt DL, Hurwitz C, et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2009;113(26):6558–66.
Fasan A, Alpermann T, Haferlach C, Grossmann V, Roller A, Kohlmann A, et al. Frequency and prognostic impact of CEBPA proximal, distal and core promoter methylation in normal karyotype AML: a study on 623 cases. PLoS One. 2013;8(2):e54365.
Fuchs O. Growth-inhibiting activity of transcription factor C/EBPalpha, its role in haematopoiesis and its tumour suppressor or oncogenic properties in leukaemias. Folia Biol. 2006;53(3):97–108.
Agrawal S, Hofmann W-K, Tidow N, Ehrich M, van den Boom D, Koschmieder S, et al. The C/EBPδ tumor suppressor is silenced by hypermethylation in acute myeloid leukemia. Blood. 2007;109(9):3895–905.
Acknowledgments
This paper forms part of Maria Kavianpour’s M.Sc. thesis. We extend special thanks to Ahvaz Jundishapur University of Medical Science, Ahvaz, Iran (Grant Number Th94/7), for the financial support. We appreciate the contribution of all our colleagues in the Health Research Institute, Research Center of Thalassemia & Hemoglobinopathy.
Authors’ contributions
Najmaldin Saki conceived the manuscript and revised it. Maria Kavianpour, Ahmad Ahmad Zadeh, and Saeid Shahrabi wrote the manuscript and prepared the tables and figure.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None
Additional information
Highlights
Detection of oncogene or tumor suppressor gene mutations has been proposed for consideration of AML prognosis.
All oncogenes and tumor suppressor gene mutations cause poor prognosis in AML patients except for C/EBPα.
Oncogene or tumor suppressor gene mutations can be used as potential MRD markers.
Rights and permissions
About this article

Cite this article
Kavianpour, M., Ahmadzadeh, A., Shahrabi, S. et al. Significance of oncogenes and tumor suppressor genes in AML prognosis. Tumor Biol. 37, 10041–10052 (2016). https://doi.org/10.1007/s13277-016-5067-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5067-1