
Abstract
As much as the cellular viability is important for the living organisms, the elimination of unnecessary or damaged cells has the opposite necessity for the maintenance of homeostasis in tissues, organs and the whole organism. Apoptosis, a type of cell death mechanism, is controlled by the interactions between several molecules and responsible for the elimination of unwanted cells from the body. Apoptosis can be triggered by intrinsically or extrinsically through death signals from the outside of the cell. Any abnormality in apoptosis process can cause various types of diseases from cancer to auto-immune diseases. Different gene families such as caspases, inhibitor of apoptosis proteins, B cell lymphoma (Bcl)-2 family of genes, tumor necrosis factor (TNF) receptor gene superfamily, or p53 gene are involved and/or collaborate in the process of apoptosis. In this review, we discuss the basic features of apoptosis and have focused on the gene families playing critical roles, activation/inactivation mechanisms, upstream/downstream effectors, and signaling pathways in apoptosis on the basis of cancer studies. In addition, novel apoptotic players such as miRNAs and sphingolipid family members in various kind of cancer are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The main goal for a cell is to stay alive during the lifetime. Holding the key of proliferation events as much as death mechanism has vital importance for the cells to keep the balance between living and death cells in the body. As one of the cellular death mechanisms, apoptosis, also known as programmed cell death, can be defined as the process of a proper death of any cell under certain or necessary conditions. Apoptosis is a part of natural homeostatic mechanism to keep the number of the cells constant in an organism and helps the tissue to eliminate increasing number of unwanted/unneeded cells that are damaged or no longer manageable during development, growth or aging [1]. It also plays crucial roles in early development and differentiation of the embryo in order to generate a full and decent organism. Although the term of “apoptosis” was firstly used by Kerr et al. in 1972 to identify a distinguished type of cell death, the first description and understanding of the programmed mechanisms of apoptosis was derived from the studies on the development of the nematode Caenorhabtidis elegans in 1999 [2, 3].
Apoptosis is a defense mechanism against damaged, stressed, or stimulated cells by any agents to prevent accumulation of non-functional cells in the tissues. If apoptosis is not be mediated properly in unwanted cells, the mutations could continue to accumulate in the cells that eventually could lead to generation of cancer and other diseases such as auto-immune diseases, AIDS, or some of neurodegenerative disorders [4]. However, apoptosis is the best defined and well-understood “programmed” type of cell death; there are many different types of cellular death mechanisms such as pyroptosis, necrosis, or autophagy, and some others might not yet be discovered [5, 6]. Apoptosis regulatory pathways, including a number of gene families, orchestrate the specific morphological and biochemical changes in the cells during the process. We touched on these significant chances briefly and focused on the genes involved in apoptosis with details.
Morphological and biochemical processes of apoptosis
Many biochemical events and a series of morphological changes occur at the early stage and increasingly continue till the end of apoptosis process. Some of the changes such as cell shrinkage, chromatin condensation, or nuclear differences could be observed by microscopic techniques [1, 7]. Morphological event cascade including cytoplasmic filament aggregation, nuclear condensation, cellular fragmentation, and plasma membrane blebbing finally results in the formation of apoptotic bodies. All the morphological hallmarks of apoptosis can be gathered under three headings; (i) the changes occur in nucleus; (ii) cell membrane and cytosolic changes; (iii) those happen in mitochondria [1, 8]. Chromatin condensation, DNA fragmentation, and nuclear fragmentation are the nuclear changes that could be observed with light and fluorescence microscopy during apoptosis. The apoptotic cell loses its association with other cells at initiation stage of the process by different signals breaking the connection. This separation followed by apoptotic body formation and resulted in blocking the inflammatory reaction of the cells, since they package their ingredient and do not release any contaminant outside the cell. Then, the buddies are rapidly phagocytized by other neighbor cells and these absorber cells do not produce any signal that causes any inflammatory response. Also, mitochondria play an important role by interacting with many different apoptotic/anti-apoptotic proteins and releasing signal molecules [9, 10].
Chromatin condensation and nuclear fragmentation are the major modifications observed in the nucleus, eventually resulting in pyknosis (chromatin condensates irreversibly that signs cell death) and followed by karyorrexis (nuclear fragmentation, the last event in nucleus during apoptosis) [11]. The fragmentation of double-stranded DNA into 180–200 bp sequences in length by the help of caspase proteins is also another essential hallmark of nuclear events during apoptosis. Caspases are responsible for DNA repair during replication as well as the termination stage of apoptosis. They are also involved in the fragmentation in apoptosis together with DNA fragmentation factors (DFFs) and endonucleases. Most of the nuclear changes of apoptosis observed by electron microscopy or even light microscopy make the apoptosis process to be determined easily [11, 12].
As soon as the apoptosis is initiated in the cells, they lose the connection between the neighboring cells; membrane shrinks and the cell packs its cytosolic ingredients into apoptotic buddies. The apoptotic buddies will be eliminated by phagocytotic cells that recognize phosphatidylserine, which is normally located in the inner side of the cell membrane and flips to the outer membrane during apoptosis [13]. At the same time, the cytoplasmic scaffold proteins and cell junction proteins such as actin, β-catenin, spectrin, or Gas2 are deactivated by cleavage and the cell loses its integrity by the function of caspases [14].
Mitochondria have complex and important roles by providing various pro-apoptotic signals, creating a downstream cascade of apoptosis activation. The balance between the pro-apoptotic and anti-apoptotic molecules keeps the cellular homeostasis stable and determines the cell fate, which is either apoptosis or proliferation. Mitochondria have a leading role in releasing a number of important apoptosis inducing molecules including cytochrome c, SMAC, apoptosis-inducing factor, or endonuclease G as a result of permeabilization of mitochondrial membrane. Permeabilization is triggered by pro-apoptotic B cell lymphoma (Bcl)-2 family proteins, while the integrity of mitochondrial membrane is maintained by anti-apoptotic members of Bcl-2 family [10, 15, 16].
Several biochemical changes such as protein modifications/degradations, DNA and chromatin deteriorations, and synthesis of cell surface markers form morphological process during apoptosis. Caspases are mainly responsible for these changes with their extensive capabilities to cleave certain molecules from one or more specific points, causing degradation and inactivation of target protein. Moreover, they can also inhibit the negative regulatory domains of specific proteins, which leads to the activation of subjected molecule [17, 18]. They are also involved in DNA fragmentation process.
Intrinsic and extrinsic apoptotic pathways
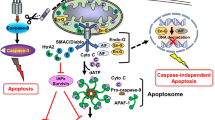
Apoptosis can be stimulated by two different pathways: (i) intrinsic pathway (or mitochondria) that mainly occurs via release of cytochrome c from the mitochondria, which activates different caspases as downstream signals, and (ii) extrinsic pathway when Fas death receptor is activated by a signal coming from the outside of the cell. After the activation of different intermediate molecules by signaling cascade, both of the pathways meet up at the final caspase activation step and commonly lead to cleavage of different proteins [19] (Fig. 1).

Two main apoptotic pathways; intrinsic and extrinsic pathways. In the extrinsic pathway, interaction between the death receptors and their ligands initiates the pathway, resulting in caspase 8 activation. This activation can be inhibited by cFLIP. Caspase 8 can directly induce apoptosis or activates caspase 3 or Bid, which lead to apoptosis. On the other hand, intrinsic pathway can be initiated by DNA damage. As a response, the cells can trigger apoptosis through mitochondrial pathway, which starts with the activation of the pro-apoptotic member of the Bcl-2 family, Bax. Anti-apoptotic proteins inhibiting the action of Bax are located on the membrane of the mitochondria such as Bcl-2 and Bcl-XL. Release cytochrome c, APAF-1 complex, and pro-caspase9 can be gathered in the cytosol, which is called apoptosome. The formation of this complex will result in the activation of caspase 9 followed by the transformation of pro-caspase-3 to caspase 3, which is the last step for apoptosis. The cross talk between extrinsic and intrinsic pathways of apoptosis is regulated by Bid, a pro-apoptotic member of Bcl-2 family. The cleavage of Bid is mediated by caspase 8, which induces apoptosis by releasing cytochrome c release from the mitochondria
Intrinsic pathway
The intrinsic pathway of apoptosis is independent from a receptor signaling, and mitochondria-associated stimuli create an intracellular signaling. The inner activation of this pathway makes the cell undergo apoptosis in either a positive or negative manner. The positive stimuli (e.g., toxic materials, viral infections, and radiations) directly activate all the mediators for apoptosis, whereas negative stimuli (loss of growth factors, different cytokines, or certain type of hormones) work in contrast to positive one and eliminate the factors that suppress apoptosis in the cells and cause apoptotic activation [1, 20]. In addition to different infections or cytokine-mediated intrinsic apoptosis activation, DNA damage also majorly induces apoptosis as a protection mechanism of the cells that do not let self to continue proliferation with an imperfect DNA sequence. DNA damage or any other type of apoptosis stimuli basically causes the changes in the trans-membrane potential of mitochondria, which result in the release of pro-apoptotic proteins into the cytoplasm.
Cytochrome c, Smad, or high-temperature requirement protein A2 (HtrA2)/Omi are a group of pro-apoptotic molecules released from mitochondria and cause the activation of caspase protein cascade [21, 22]. Cytochrome c interacts with Apaf-1, resulting in the formation of “apoptosome” complex, which activates pro-caspase-9. After active caspase-9 activates caspase-3, the final cascade is become activated and nucleus will be fragmented together with the breaking of nuclear membrane [23]. This stage is the initial event for extrinsic and intrinsic pathways of apoptosis, where caspase-3 cleaves the different proteins such as kinases, DNA control proteins, cytoskeletal proteins, or inhibitor of endonucleases. DNA condensation, membrane blebbing, and all the morphological changes are regulated by caspases as a common mechanism for both intrinsic and extrinsic trigger [17].
On the other hand, another group of molecules released by mitochondria including endonuclease G or AIF are also pro-apoptotic proteins but involved in the process at the later stages. These molecules are trans-located into the nucleus where they first cause an elementarily DNA fragmentation and chromatin condensation which is defined as “stage 1,” and an advanced condensation and DNA fragmentation by the help of caspase-3 at later stage is called as “stage 2” [24].
All the intrinsic apoptosis events are primarily controlled by Bcl-2 family of proteins and p53 tumor suppressor protein which is majorly involved in the activation of Bcl-2 family proteins. The members of Bcl-2 protein family can act as either pro-apoptotic (Bax, Bak, Bid, Bim, Puma, Noxa, Bad, and Blk) or anti-apoptotic (Bcl-2, Bcl-XL, Bcl-X, and BAG) and also determine the membrane integrity of mitochondria and are involved in the process of cytochrome c release [1, 25].
Extrinsic pathway
Apoptosis triggered by extrinsic pathway is primarily mediated by signaling through membrane-bound death receptors that belong to tumor necrosis factor (TNF) gene superfamily. The initial signal is provided by the interactions between the ligands and cell membrane death receptors such as Fas ligand/FasR, TNF/TNF R1, Apo2L/DR4, or TNF-related apoptosis-inducing ligand (TRAIL) R1, which is resulted in ligation of death domains of these receptors [26]. Binding of Fas ligand to its receptor induces the binding of adaptor protein, Fas-associated death domain (FADD), while TNF/tumor necrosis factor receptor (TNFR) interaction causes the binding of TNFR-associated death domain (TRADD), which is resulted in pro-caspase-8 activation. Pro-caspase-8 is activated auto-catalytically by the help of death-inducing signaling complex (DISC). Active caspase-8 either induce Bid, thus intrinsic pathway also become involved and activated with an outside signal, or caspase-3 and caspase-7 and the activation process of apoptosis is terminated with the same final pathway as intrinsic stimuli does [1, 19, 27, 28]. Bid is the pro-apoptotic member of Bcl-2 family, exhibiting a common molecule between intrinsic and extrinsic pathways of apoptosis. Caspase-8 causes the cleavage and myristoylation of cytoplasmic Bid protein, leading to its movement through mitochondria. Then, apoptosome formation is induced by cytochrome release via Bak and Bax molecules [29].
The extrinsic activation of apoptosis can also be inhibited via two different ways. The one is binding of FLICE-like inhibitory protein (cFLIP) to FADD and pro-capase-8 and blocking their activity, and the other way is inhibition of caspase-8 biogenesis by a protein named Toso which is firstly described in T cells [30, 31].
In the next sessions, we will discuss all the gene families involved in intrinsic or extrinsic pathways of apoptosis with their main player and their functions.
Caspase family members
Caspase family comprise conserved cysteine aspartic-specific proteases, and members of caspase family are considerably crucial in the regulation of apoptosis [32]. In C. elegans, Ced-3 was reported to be essential for the cell death, which is mainly conducted by the caspase Ced-3, Ced-4 activating Ced-3, and Ced-9 inhibiting apoptosis [33, 34]. There are 14 different caspases in mammals, and they are basically classified as the initiators including caspase-2, -8, -9, and -10; and the effectors including caspase-3, -6, -7, and -14; and also the cytokine activators including caspase-1, -4, -5, -11, -12, and -13 [35, 36]. Structurally, while initiator caspases have long N-terminal pro-domain known as caspase recruitment domains (CARDs) including more than 90 amino acids, effector caspases have shorter sequences known as death effector domain (DED) including 20–30 amino acids. Since caspases firstly synthesized as zymogens, they are subsequently activated during the apoptotic process. While initiator caspases are self-activated, effector caspases are activated by initiator caspases via internal cleavages [37]. Rather than apoptosis, most of caspase family members are functional in cellular proliferation, survival, and inflammation, whereas some of them are essential for apoptosis [38, 39].
Caspase-1, the first identified caspase, is interleukin-1b processing enzyme (ICE), and it is known as Ced-3 homologue [40]. Caspase-1 is involved in cytokine activator group of caspase family since inflammatory cytokines, pro-IL-1b and pro-IL-18, are the main substrates for caspase-1 [39]. While caspase-1 is not essential for apoptotic signaling, it is essential in inflammation process [41].
The second identified caspase, caspase-2, containing CARD, plays important roles in DNA damage-, metabolic abnormality-, and ER stress-induced apoptosis [42]. Caspase-2 is known as a substrate for both caspase-3 and caspase-8 [43, 44]. Caspase-2 activation comprises the formation of PIDDosome complex including RIP-associated ICH-1/ECD3 homologous protein with death domain (RAIDD) that have CARD and death domain (DD) and p53-induced protein with death domain (PIDD) [45]. Functional properties of caspase-2 have still not been clarified thoroughly.
Caspases-3, -6, and -7 are involved in the effector caspase group, and they act in a similar manner in the apoptotic process [46]. Caspase-3 is activated via both extrinsic and intrinsic apoptotic pathways [47]. Despite there are limited information about caspase-6 and -7 rather than caspase-3, it is known that while caspase-3 suppression results in the inhibition of apoptosis, suppression of caspase-6 and -7 do not significantly affect the apoptotic process [48]. Furthermore, caspase-3 was reported to be crucial for PARP cleavage and DNA fragmentation which are hallmarks of apoptosis [49]. However, some studies showed that under the conditions that both caspase-3 and caspase-7 were knocked out, the cells could undergo cell death in an alternative manner via necrosis. Studies with caspase-3/caspase-7 double-knockout thymocytes and mouse embryonic fibroblasts showed that thymocytes remain sensitive to Fas-mediated apoptosis, whereas fibroblasts become resistant [49, 50].
Functionally well-known member of caspase family, caspase-8, is crucial factor for TNF-induced extrinsic apoptotic pathway [51]. Pro-caspase-8 is recruited to DISC by FADD, and dimerization or trimerization triggers pro-caspase-8 activation via reciprocal cleavage. Caspase-8, in turn, cleaves and activates caspase-3, -7, Bid, and also NF-kB [52, 53]. Caspase-8 activation is regulated by cFLIP that is structurally homologous to caspase-8 but does not have caspase activity [54]. FLIPS, the short isoform of cFLIP, controls the DISC formation in a negative manner. However, the long isoform of cFLIP, FLIPL, has reciprocal effect on DISC formation and caspase-8 activity. While some studies reported FLIPL to be inducer of DISC formation, some reports showed that it could be an inhibitor [55, 56]. Inhibitor and inducer effects of FLIPL on caspase-8 activity depend on FLIPL levels. At low concentrations, generation of heterodimers between FLIPL and pro-caspase-8 or pro-caspase-10 induces their activity. However, at higher levels of FLIPL, caspase-8 activation diminishes and NF-kB activation increases [57, 58].
Caspase-9, the initiator caspase, is an important factor for the generation of apoptosome complex in the mitochondrial pathway. Once cytochrome c is released from the mitochondria, it binds to Apaf1 which is the receptor for cytochrome c in the cytoplasm [59]. Cytochrome-c and Apaf-1 generate apoptosome, and then, pro-caspase-9 binds to Apaf-1. Afterward, pro-caspase-9 is activated via reciprocal cleavage, and by this way, apoptosome complex also become activated. Then, caspase-3 is cleaved and activated via caspase-9 found in the active apoptosome complex [60].
Inhibitors of apoptosis proteins and inhibitors of apoptosis protein antagonists
Inhibitors of apoptosis proteins (IAPs) were firstly discovered in Baculovirus as gene products. All IAPs have baculovirus IAP repeats (BIRs) that composed of one or more zinc finger motifs [61]. The first identified IAP, OpIAP, inhibits pro-caspase cleavage and activation rather than direct inhibition of caspase activity [62]. NAIP, the first identified mammalian IAP, was reported to be related to the generation of immune response against bacterial infection. Hence, it is not directly correlated with caspase inhibition [63].
Survivin/BIRC5, another identified mammalian IAP, was firstly reported as caspase inhibitor, but now, it is known that survivin does not directly inhibit caspase activity. Mechanism of action of survivin, bearing one BIR domain, is to assemble with centromeres and p21Waf1 at the beginning of mitosis [64, 65].
XIAP/BIRC4, the mostly clarified mammalian IAP, has three BIR domains and a RING domain; it is located on X chromosome; and also, it is quite effective in apoptosis inhibition via inhibiting caspase activity [66]. Mainly, caspase-3 and caspase-7 can be inhibited by XIAP via inserting a residue of aspartic acid into the caspase active region [67]. Additionally, XIAP can also inhibit caspase-9 activity via binding the third BIR to the N-terminus of pro-caspase-9, resulting in the prevention of caspase-9 dimerization [68].
Similarly, another mammalian IAPs, cIAP1/BIRC2, and cIAP2/BIRC3 have three BIR domains and a RING domain in their structures [63]. Rather than inhibiting directly caspase activity, they inhibit apoptosis indirectly. They can bind IAP inhibitor, second mitochondrion-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (SMAC/DIABLO), trigger NF-kB and MAPK activity, and also they can trigger proteasomal degradation of caspases [69].
Bruce/BIRC6, a member of IAP family, is mainly found in secretory organs, testis, lymphatic cells, and brain. Similar to survivin, Bruce located at the outer membrane of the trans-Golgi network bears only one BIR domain. Bruce can inhibit caspase-3, -6, -7, -8, and -9, and also, it can trigger the proteasomal degradation of SMAC/DIABLO. Many studies showed that Bruce is upregulated in ovarian and brain cancer cell lines, resulting in development of resistance against apoptotic agent [70].
IAP antagonists, SMAC/DIABLO, HtrA2/Omi, and XIAP-associated factor 1 (XAF1) are known as potent inhibitors of IAPs. SMAC/DIABLO bears a mitochondrial targeting signal (MTS) at N-teminus, and it become mature after the MTS cleavage [21]. Once apoptosis is triggered, SMAC/DIABLO is delivered into the cytosol, and SMAC/DIABLO homodimers bind to IAPs via its Ala-Val-Pro-Ile sequence in N-terminal domain [71]. Interaction of SMAC/DIABLO with XIAP via the second and third BIRs results in caspase-3 and caspase-9 release [72]. It was reported that SMAC is overexpressed in several types of solid tumors such as colon, stomach, prostate, ovary, and lung cancers [73]. Likewise, HtrA2/Omi delivery is triggered via apoptotic induction and then, it binds to IAPs through its IAP-binding motif (IBM) [74]. XAF1, the other potent IAP antagonist, binds to BIR domains of IAPs such as XIAP, cIAP1, and cIAP2 and by this way, promotes apoptosis [75]. A study suggesting XAF1 as a prognostic marker for colon cancer showed that XAF1 is overexpressed in colon cancer cells as compared to adenoma cells which are benign [76].
Bcl-2 family members
Bcl-2 family members, which play important roles in regulating apoptotic signaling, are divided into three subfamilies including (i) pro-survival subfamily members (Bcl-2, BclXL, BclW, MCL1, and BFL1/A1), (ii) BH3-only subfamily members (Bad, Bim, Noxa, and Puma9), and (iii) pro-apoptotic mediator subfamily members (Bax and Bak) [77, 78]. Basically, all of the members of Bcl-2 family share typical characteristic functions; (i) they dimerize with other members of Bcl-2 family, (ii) they contribute to the regulation of mitochondrial homeostasis by binding proteins, and (iii) they contribute to outer mitochondrial membrane pore formation [79].
The members of BH3-only subfamily members become activated under stress conditions like growth factor deprivation and DNA damage. Active BH3-only proteins, in turn, inactivate members of pro-survival subfamily via binding that promotes the activation of the members of pro-apoptotic subfamily members. Active pro-apoptotic subfamily members, Bak and Bax, then provide cytochrome c release from the mitochondria through permeabilizing the outer mitochondrial membrane [80].
In several types of human malignancies, the balance between the expression levels of Bcl-2 family genes is broken down, and the equilibrium changes to the pro-survival subfamily member direction. In this case, cancer cells can escape from apoptotic signals and therefore develop resistance against therapeutic agents [81, 82]. Additionally, Bcl-2 family members are also considered in cancer therapy due to their therapeutic potentials [83]. In the clinical trials, the BH3-only mimetic agents targeting Bcl-2 are being investigated in order to find alternative potent therapeutic approaches in several types of malignancies [84–86].
TNF gene superfamily
One of the most important ways of triggering apoptosis is mediated through death receptors (DRs), which are classified in TNF superfamily including also ligands such as TNF, TRAIL, and Fas ligand (FasL) [87]. The induction of apoptosis by these ligands is initiated by binding to their specific membrane receptors [88, 89]. TNF superfamily is known to comprise 19 ligands and 29 receptors that function in highly different processes in the body including inflammation, apoptosis, proliferation, and invasion [90]. Even though all members of TNFR superfamily are generally trimeric type I trans-membrane proteins and possess cysteine-rich extracellular subdomains, they are actually different in their primary structure, which make them unique to recognize their ligand in a specific and exclusive manner [91]. These DRs also contain a homologous cytoplasmic cysteine-rich “DD” which is responsible for transmission of apoptotic signals from cell surface to intracellular signaling pathways [92]. Because, adapter molecules such as FADD and TRADD have these death domains as well to interact with DRs. The ligands included in TNF superfamily share a common extracellular TNF homology domain (THD), which is involved in the formation of homotrimers via non-covalent bonding [93]. The extracellular domain of most TNF ligands undergoes proteolytic cleavage to make a soluble ligand, even though they are synthesized as type II trans-membrane proteins [93].
CD95 (DR2/Fas/APO-1), TNF receptor 1 (DR1/TNFR1), TRAIL-R1 (DR4), and TRAIL-R2 (DR5) are the best characterized DRs of which ligands are CD95 ligand (CD95L/FasL), TNFα, lymphotoxin-α (these two bind to TNFRI), and TRAIL (these two bind to TRAIL-R1 and TRAIL-R2), respectively [94]. The genes encoding TNF superfamily receptors and ligands are the scope of this part together with their significant contribution to apoptosis specifically.
Fas cell surface death receptor and Fas ligand
The proteins encoded by Fas cell surface death receptor (FAS) and Fas ligand (FASLG) genes are members of the TNF superfamily. Fas also known as CD95/APO-1/DR2 is one of the best studied DRs with molecular weight of 48 kDa. Fas gene occupies about 25 kb on human chromosome 10 with nine exons in which exon 6 codes for trans-membrane domain [95]. This receptor containing a DD plays a crucial role in the regulation of programmed cell death and is involved in the pathogenesis of various malignancies such as cancer. On the other hand, FasLG gene is located on human chromosome 1, which encodes a type II trans-membrane protein called FasL (CD95L) present at the surface of activated immune cells such as T cells and natural killer cells [93]. Therefore, Fas/FasL interaction results in the elimination of infected and transformed cells, which is generally used by immune cells to avoid cancer development.
The interaction of Fas with FasL results in changes in the conformation and aggregation of receptor on the plasma membrane and triggers an initial signaling event through protein-protein interactions. The main structural changes take place in DD of receptor, which recruits FADD through its DD. Then, FADD interacts with pro-caspase-8 and -10. After activation of these caspases via auto-cleavage, they are released in the cytosol as active caspases resulting in the apoptotic cell death. The complex CD95/FADD/caspase-8/-10 is called DISC stands for “death-inducing signaling complex” [96].
In the literature, there are various kinds of studies identifying the roles of Fas/FasL in order to induce apoptosis in several malignancies especially in cancer. Escaping from apoptotic stimuli is a very well known feature of cancer cells which might develop multiple strategies to inhibit apoptotis mediated by CD95. In a majority of cancer types from different origins, somatic mutations in CD95 gene were found to be a common way to trigger the development of resistance toward apoptosis. For instance, 5′ region of CD95 gene was analyzed in terms of somatic mutations in nodal diffuse large B cell lymphoma and defined mutations were shown to be included in progression of diseases due to inhibition of apoptosis [97]. Another common way to become resistant to CD95-induced apoptosis is the regulation of surface expression of receptor [98]. In a study performed by Ivanow et al. (2006) [98], the treatment of melanoma cells having increased surface expression of Fas receptor with soluble FasL resulted in the induction of apoptosis. CD95-mediated apoptosis can also be blocked by decreasing expression of FADD or caspase-8 [99, 100].
In a recent study, the effect of a toxic steroid on human bladder cancer cells was found to be related to increased expressions of Fas and FasL in vitro and in vivo at both messenger RNA (mRNA) and protein levels [101]. Zhong et al. (2015) [102] showed that the mechanism of resistance to Fas-mediated apoptosis in human hepatocellular carcinoma cells (HCCs) and overexpression of oxysterol-binding protein-related protein 8 (ORP8) which is normally downregulated in HCC was found to induce apoptosis by upregulating FasL.
TRAIL
TRAIL (also called as APO2 ligand) is a type II trans-membrane protein and processed proteolytically at the cell surface to form a soluble ligand. TRAIL is grouped into the TNF cytokine family, which was discovered based on its extracellular domain sequence homology with CD95L and TNF [103]. TRAIL-mediated apoptosis takes place after its binding to its DD containing receptors, TRAIL receptor 1 (death receptor 4, DR4), and TRAIL receptor 2 (death receptor 5, DR5) [104]. There are also three other TRAIL receptors, which do not possess apoptotic ability and function as decoys. Decoy receptors 1 (DcR1) and 2 (DcR2) are expressed on the cell surface similar to DR4 and DR5. Even though their extracellular and ligand-binding domains show significant homology to DR4 and DR5 and are fully functional, they lack of functional intracellular DD [105]. Therefore, increased expression of either DcR1 or DcR2 provides resistance against TRAIL-induced apoptosis [104]. In a recent study, it was shown that coexpression of DcR1 and DcR2 with DR4/DR5 on the same cell can block apoptosis. However, TRAIL was engineered in order to escape from binding to DcRs, which were found to still exert trans-cellular regulation originating from stromal cells and affect tumor cells. Therefore, it is important to target these decoy receptors selectively to gain maximum efficacy [106]. Another recent study showed that DR4 and DR5 were upregulated while DcR1 and DcR2 downregulated in colon cancer cells after their treatment with a non-steroidal anti-inflammatory drug. Therefore, colon cancer cells became sensitive to TRAIL-induced apoptosis [107]. The fifth TRAIL receptor is osteoprotegerin (OPG), which is a secreted receptor for TRAIL with low affinity. OPG has sequence homology to TNF receptor superfamily, however but lacks of a trans-membrane domain. OPG can bind to TRAIL and prevents its interaction with death receptors, thus preventing Jurkat cells from undergoing TRAIL-mediated apoptosis [108]. Therefore, OPG has an anti-apoptotic effect by preventing interaction between TRAIL and the death receptors [109]. Lane et al. (2012) [110] showed the relationship between higher OPG expression and resistance of ovarian cancer cells to TRAIL-induced apoptosis.
Similar to apoptosis induction by Fas/FasL, binding of TRAIL to its DRs, DR4 and DR5 recruits the adaptor protein FADD and inactive versions of caspase-8 and -10 to form the DISC complex in which caspase-8 and -10 become activated by auto-proteolytic cleavage and released in the cytosol to activate effector caspases [111].
The size of human TRAIL gene is ~20 kb, containing five exons and four introns. The first exon encodes for the trans-membrane domain and the cytoplasmic domain, whereas exons 4 and 5 code for the extracellular domain responsible for the interaction of TRAIL with its receptors. Exon 5 also encodes for the C-terminal amino acids along with containing the 3′-untranslated region (3′-UTR) and poly-A tail [112]. There are various variants of TRAIL; however, only one specific isoform of TRAIL (TRAILα) is responsible for its cancer-selective apoptosis induction potential [113].
TRAIL gene is subjected to strict regulation due to its critical role in the induction apoptosis. Altered expression of TRAIL gene has been found in various kind of disease. Multiple studies have been performed to analyze single-nucleotide polymorphisms (SNPs) in various patient populations. In a recent study, peripheral blood samples were analyzed in terms of SNPs in TRAIL promoter and substitution of C to T at position -723 was found to be significantly associated with sporadic breast cancer and decreased TRAIL mRNA levels due to transcriptional repression [114]. Bos et al. (2009) found that TRAIL mRNA expression decreased in breast cancer patients with brain metastasis [115].
TRAIL has been considered as a potential therapeutic target due to its selective apoptosis inducing action in cancer cells as compared normal cells [116]. There are several ongoing and completed clinical phase studies targeting TRAIL apoptotic pathway by using agents including monoclonal antibodies and recombinant human proteins, and these studies are giving promising results with no significant toxic effects [117].
TNF and TNF receptor 1–2 (TNFR1 AND TNFR2)
TNF is expressed as a trans-membrane 26-kDa protein and then undergoes proteolytic cleavage to form 17-kDa trimeric soluble cytokine. The resulting TNF functions by binding to two different receptors, TNRF1 and TNFR2, which are also trimeric trans-membrane proteins [90]. TNF is produced by immune cells including activated natural killer cells, T cells, and activated monocytes/macrophages and a wide range of non-immune cells such as fibroblasts [118]. TNF/TNFR signaling is well known to be involved in various cellular functions such as apoptosis, cell proliferation, and differentiation [119].
TNF-mediated apoptosis is generally carried out by TNFR1 (DR1), since only TNFR1 is known to contain DD [87]. Binding of TNF to TNFR1 triggers the recruitment of TRADD protein through its DD. Then, TRADD interacts with FADD resulting in the recruitment of pro-caspase-8, which is proteolytically cleaved to active caspase-8. Caspase-8 then activates caspase-3 responsible for apoptotic cell death [120]. The activity of caspase-8 is strictly regulated by a negative inhibitor protein cFLIP that contains DED instead of DD. cFLIP interacts with pro-caspase-8 to prevent its continuous recruitment to the TNFR1 DISC [121]. TNF-induced cell death occurs only under stress conditions such as altered cell metabolism, inhibition of cell cycle progression, and protein synthesis [120]. Therefore, it only induces apoptosis in transformed cells (cancer cells), virus-infected cells, or stressed cells, not in normal healthy cells. Interestingly, TNFR2 has been shown to function in cell proliferation unlike TNFR1 [122].
In a recent study, IL32-α, a novel cytokine, was found to inhibit colon cancer cell growth in an experimentally generated colon cancer model by increasing TNFR1-induced cell death signaling, which was evidenced by increased expression of TNFR1. Yu et al. [123, 124] found a new way of apoptosis induction in adenosine-treated colon cancer cells, which included increased expression of TNFR1. TNFα secretion increased in acute myeloid leukemia cells after treatment with SMAC mimetic and IFNα combination [125]. Inhibition of TNFα and TNFR1 by a pharmacological inhibitor and genetic silencing, respectively, reduced SMAC mimetic/IFNα-triggered apoptosis. Tao et al. suggested that survivin, an inhibitor of apoptosis, inhibitor induced programmed cell death in Wilms tumor cells by increasing expression of TNFR1 signaling [126].
As well as TNFα, lymphotoxin α (LTα) which is secreted as a homotrimer binds to TNRF1 [127]. Even though both TNFα and LTα bind signal via TNFR1, LTα was found to possess less ability to induce TNRF-mediated cell death in an early study [128]. However, a recent research by Etemadi et al. displayed that LTα has same potential to induce apoptosis via TNFR1 signaling [129].
Genetic changes in the promoter region of TNFα have been well studied in several human diseases to find correlation between apoptosis induction and TNFα expression. De Oliveria et al. studied the effect of a polymorphism, TNF-α-857 C/T, on gastric cancer patients, found that this mutation decreased mRNA level of TNFα resulting in resistance to apoptosis [130].
Death receptor 3
Death receptor 3, also known as APO-3, has four characteristic cysteine-rich motifs with molecular weight of 53.5 kDa. Death receptor 3 (DR3) gene is localized on chromosome 1 and encodes a type II trans-membrane protein. Similar to other TNFR family members, DR3 has a DD in its cytoplasmic part and initiates apoptotic signaling [87].
Most of the studies have been related to DR3 involvement in immune system modulation due to its frequent expression on lymphoid tissues such as the spleen, thymus, and peripheral blood lymphocytes [131]. The role of DR3 in the development of some human malignancies has also been enlightened. DR3 expression was shown to be increased in human colon cancer cells treated with cordycepin, a deoxy form of adenosine, resulting in apoptosis induction [132]. A specific phenolic compound triggered apoptosis in human non-small-cell lung cancer (NSCLC) cells by increasing the expression of DR3 [133]. Silencing of DR3 via siRNA approach reversed its growth inhibitory effect.
p53
p53 is encoded by human TP53 gene localized on the short arm of chromosome 17 with a molecular mass of 43.7 kDa [134]. It occupies 19.200 bp including 11 exons. There are various p53 isoforms based on alternative splicing of TP53 gene; some of which play opposite roles as compared to p53, while others have similar functions like full-length p53 [135].
Human p53 protein is composed of the following three different domains with important functions: the DNA-binding domain, the N-terminal trans-activational domain, and the C-terminal oligomerization domain [136]. DNA-binding domain binds to response elements of target genes, whereas the N-terminal trans-activation domain forms binding sites for several negative or positive regulators. The C-terminal domain undergoes alternative splicing and posttranslational modifications [137].
p53 acts as tetrameric transcription factor, which controls the expression of a large set of genes involved in significant cellular processes including DNA damage detection, cell cycle arrest, apoptosis, DNA repair, and senescence [134]. It is both involved in intrinsic and extrinsic pathways of apoptosis by inducing transcription of several proteins like PUMA, Bid, Bax, TRAILR2, and CD95, which is called transcription-dependent apoptotic pathway of p53 taking place in the nucleus [138]. In this pathway of p53-dependent apoptosis, the trans-activation domain of p53 interacts with the players of basal transcription machinery such as the transcriptional coactivator p300/CBP [139]. In normal healthy cells, p53 levels are very low due to its rapid turnover; however, if a damage is detected in the cells, p53 becomes stabilized resulting increased p53 level [140]. p53 stability is controlled by mouse double minute 2 (MDM2) gene, which is an E3 ubiquitin ligase that negatively regulates p53 stability through ubiquitination, thus proteosomal degradation. MDM2 also inhibits the interaction between the p53 trans-activation domain and the components of transcription machinery [141]. MDM2 has been found to be overexpressed in many cancer cells, which leads to neutralization of interaction between p53 and transcription machinery components, thus impairing transcription-dependent apoptosis [142, 143].
p53 has the ability to activate intrinsic pathway of apoptosis by inducing the transcription of especially apoptotic Bcl-2 family genes such as PUMA [144]. p53 induces PUMA mRNA expression immediately in response to DNA damage by binding the two p53-responsive elements in the PUMA promoter. p53 binding results in the acetylation of core histones, H3 and H4, which is responsible for chromosome decondensation and transcriptional activation [145]. Similarly, TRAIL-R2 expression is induced by p53, which binds to a p53-responsive element in TRAIL-R2 promoter in order to induce extrinsic pathway of apoptosis [140]. As an alternative mechanism, p53 functions a transcriptional repressor of certain anti-apoptotic genes including survivin which promotes caspase activation [146]. p53 is also directly involved in apoptosome formation by activating transcription of Apaf-1 gene including a p53 response element in its promoter [54]. In response to DNA damage, p53 is also displayed to activate caspase-6 cleaving nuclear envelope protein lamin A and various transcription factors through a response element within the third intron of the gene [147].
As well as transcription-dependent functions of p53, its transcription-independent functions in terms of apoptosis have been defined. p53 induces apoptosis by acting directly at mitochondria. p53 trans-locates to the mitochondria in response to apoptotic signal, where it forms inhibitory complexes with Bcl-XL and Bcl-2 causing the permeabilization of the mitochondrial membrane and cytochrome c release [148]. The interaction between p53 and Bcl-XL and Bcl-2 is mediated by p53 trans-activation domain like p300/CBP binding, even though these two modes of p53-dependent apoptosis induction occur in different cellular compartments [149]. Moreover, cytosolic p53 might induce the activation of pro-apoptotic Bax via direct protein-protein interactions [150]. Leu et al. displayed that p53 interacts with pro-apoptotic mitochondrial membrane protein Bak, which makes Bak undergo oligomerization and releases cytochrome c from mitochondria. Binding of p53 to Bak damages interaction between Bak and anti-apoptotic Mcl-1 [151].
Mutations in p53 gene have been considered most common genetic changes in cancer. Mutant p53 proteins can both lose their native tumor suppressor activity and provide active tumor development [152]. Missense mutations form the majority of alterations in p53 gene, which commonly occur in the DNA binding domain of p53, thus preventing p53 from promoting target gene expression [153]. Recently, Saleem et al. analyzed loss of function mutations in p53 gene in the patients of oral squamous cell carcinoma and found that AGT to ACT missense mutation in DNA binding domain may result in impaired p53 function. In chronic lymphocytic leukemia patients, missense mutations in DNA binding motif were correlated with poor survival [154, 155].
p53 has been considered as a significant player of apoptosis in many studies, and there is a growing accumulation of articles revealing its role in many malignancies. In a recent study by Wang et al., acute pro-myelocytic leukemia cells were subjected to apoptosis when treated with combination of tetraarsenictetrasulfide and arsenic trioxide through upregulation of p53 and its target gene Bax. Transcription-independent role of p53 in apoptosis induction was identified in a study in which p53 trans-located to mitochondria and induced mitochondrial membrane depolarization in HUVEC cells exposed to heat stress [156, 157].
There is an increasing attention to develop different strategies that can modulate p53-dependent apoptotic pathways such as inhibition of p53-MDM2 interaction using MDM2 inhibitors, restoring mutated p53 back to its wild-type form and p53 vaccines [158–160].
MicroRNAs in extrinsic and intrinsic apoptotic pathways
Changes in the apoptotic response in cancer can result in tumor initiation, progression, and treatment resistance [3]. There are numerous studies including the roles of microRNAs in the control of apoptosis, and these microRNAs display their effects by directly targeting genes involved in both extrinsic and intrinsic pathways of apoptosis [161]. These microRNAs (miRNAs) can be classified as oncogenic and tumor suppressive miRNAs [162]. One of these miRNAs is miR-130a that was found to reduce drug resistance in non-small-cell lung cancer by targeting MET proto-oncogene and to sensitize this cancer cells to TRAIL-induced apoptosis by inhibiting miR-221 and miR-222, which are upregulated by MET and involved in TRAIL resistance [163]. Therefore, miR-221 and miR-222 are oncogenic miRNAs that induce drug resistance and block apoptosis in several cancer types such as gliomas [164]. Another oncogenic miRNA exerting its anti-apoptotic effects by inhibiting FasL directly is miR-21, which is found to be upregulated in advanced pancreatic cancer patients [165]. miR-24 regulates apoptosis by binding to coding sequence of Fas-associated factor 1 (FAF1) mRNA and induced apoptosis of several different types of cancer [166]. miR-21 also negatively regulates PTEN, a tumor suppressor gene, involved in the apoptotic pathway through the formation of DISC complex in many tumors such as breast and gastric cancers [167, 168]. In gastric cells, miR-21 upregulated and decreased PTEN expression, resulting in significant suppression of trastuzumab-induced apoptosis [168]. miR-200c sensitized cells to apoptosis by directly targeting Fas-associated phosphatase-1 (FAP-1), which is an inhibitor of Fas-induced apoptosis [169]. miRNA-886-5p inhibited apoptosis of human cervical cancer cells by downregulating Bax [170]. Zhou et al. (2010) [171] found that miR-125b was upregulated in taxol-resistant breast cancer cells and suppressed apoptosis. Bak1, pro-apoptotic Bcl-2 antagonist killer 1, was identified as the direct target of miR-125b. miR-25 and miR-32 could directly target the pro-apoptotic function of Bim, therefore suppressing apoptosis in ovarian and human myeloid leukemia cells, respectively [172, 173]. In addition, miR-483-3p might target PUMA, whose enforced expression protects cells from apoptosis [174]. miR-34 decreases the expression of Bcl-2, resulting in an increase in apoptosis [175]. In this study, downregulation of miR-34 induced cell proliferation and invasion in malignant mesothelioma. miR-15b/16 could downregulate Bcl-2, thereby triggering apoptosis [176]. miR-153 induced apoptosis of glioblastoma cells by targeting 3′-UTR of Bcl-2 and Mcl-1 [177]. miR-491 could induce apoptosis in colorectal cancer cells by downregulating anti-apoptotic Bcl-XL [178]. There are several studies showing the roles of miRNAs in the regulation of caspase expression. miR-23a and miR-24a blocked mitochondrial apoptosis by inhibiting the expression of caspase-9 [179, 180]. The downregulation of miR-23a increased the 5-FU-induced apoptosis in colon cancer cells [179]. Wu et al. (2014) [181] displayed that miR-421 upregulated in human gastric cell lines and tissues. miR-421 downregulated the expression of caspase-3 and blocked the apoptosis of cancer cells. miR-106b-25 was shown to be upregulated in human prostate cancer and functions by partly inhibiting of caspase-7 expression [182]. Floyd et al. (2014) [183] demonstrated that miR-582-5p and miR-363 inhibit apoptosis by directly targeting caspase-3 and caspase-9 in glioblastoma. Caspase-3 has been shown to be a target of miR let-7a in human squamous carcinoma cells and hepatocellular carcinoma cells [184]. Curcumin induced apoptosis of NSCLC by the downregulation of miR-186, whose direct target is caspase-10 [185]. Based on all this evidence discussed in this particular review, miRNAs play key regulatory roles in apoptosis and could be important therapeutic targets in cancer (Fig. 2).

miRNAs involved in the apoptotic pathways. Some of the miRNAs can inhibit apoptosis by targeting the death-receptor pathway including miR-21, miR-24, and miR-200c. In the mitochondrial pathway, various miRNAs could target Bcl-2 family proteins and caspases including miRNA-886-5p, miR-125b, miR-25, miR-32, miR-483-3p, miR-34, miR-15b/16, miR-153, miR-491, miR-23a and miR-24a, miR-421, miR-106b-25, miR-582-5p and miR-363, miR let-7a, and miR-186
Bioactive sphingolipids in apoptotic pathways
Bioactive sphingolipids are a family of membrane lipids that have many regulatory roles in several cellular events such as cell proliferation, senescence, adhesion, migration, and also apoptosis [186]. Ceramide, the central molecule of bioactive sphingolipid metabolism, was reported as a regulator of apoptosis in many studies. Treatment of cancer cells with radiation and chemotherapeutics such as vincristine, daunorubicin, gemcitabine, and etoposide result in ceramide accumulation in the cells as a secondary effect of these therapeutics [187]. Decreased ceramide levels result in the development of drug resistance in cancer cells [188]. In vitro studies indicated that treatment of cancer cells with ceramide triggers the release of cytochrome-c from the mitochondria [189]. Additionally, it was reported that anti-apoptotic Bcl-2 blocks ceramide channels in an independent manner from Bak and Bax [190]. Furthermore, ceramide also induces Bax-mediated apoptosis in several types of cancer including, breast, prostate, and colon cancers [191]. Ceramide was reported that it activates cathepsin D, an inducer of apoptosis in a form of lysosomal aspartyl protease, in response to gemcitabine treatment [192]. Unlike ceramide, the other member of sphingolipid family, sphingosine 1-P (S1P) is related to cell proliferation, survival, and also inhibition of apoptosis [193]. S1P was reported to cause angiogenesis by VEGF signaling, which then triggers RAS and MAPK signaling in cancer cells. By this way, S1P causes cytoskeleton reconstruction and apoptosis inhibition [194]. While increased ceramide levels cause induction of apoptosis, increased S1P levels cause inhibition of apoptosis [195]. S1P leads to drug resistance due to its anti-apoptotic function. Sphingosine kinase-1 (SK1), which catalyzes the conversion of apoptotic ceramide to anti-apoptotic S1P, was also reported to decrease apoptotic effects of chemotherapeutic agents in prostate cancer [196]. Like S1P, glucosylceramide (GC), the other important member of sphingolipid family, is also known as a powerful anti-apoptotic molecule [193]. Glucosylceramide synthase (GCS), the enzyme catalyzing the conversion of apoptotic ceramide to anti-apoptotic GC, was found to be increased in drug-resistant cancer cells [193]. Treatment of pancreatic cancer cells with a ceramide analogue causes the accumulation of ceramide in the mitochondria, and by this way, ceramide reduces drug resistance and triggers apoptosis [197]. Additionally, treatment of pancreatic cancer cells with ceramide in combination with a GCS inhibitor, PDMP, decreases tumor growth in vivo [198]. Moreover, GC degradation increases ceramide generation resulting in the trigger of apoptosis and also reduced tumor growth in melanoma xenografts [199]. Reduced tumor growth was also reported in mouse models with breast cancer treated with C6:ceramide [200]. Furthermore, a type of sphingosine kinase inhibitor, SK2, was reported to trigger apoptosis, inhibit cell proliferation, and decrease tumor size in mouse models bearing hepatoma, mammary adenocarcinoma, and kidney carcinoma [201, 202]. Safingol, the first agent used in the clinic due to its ability in sphingosine kinase inhibition, increases apoptotic effects of chemotherapeutics used in cancer therapy [203]. An FDA-approved drug that has inhibitory effects on SK1 and SK2 activities, FTY720, inhibits cell proliferation and leads to apoptosis in mice bearing breast cancer or melanoma [204]. In chronic myeloid leukemia (CML) cell lines, increase in serine palmitoyltransferase levels via BCR/ABL inhibition result in activation of apoptotic signals [205]. GCS inhibition in CML cell lines bearing T315I mutation results in apoptosis via activating GSK-3 [206]. Another study reported that ceramide triggers apoptosis via activating p38, caspase-8, and c-Jun N-terminal kinase (JNK) in K562 CML cells [207]. Many studies from our laboratory also showed effects of bioactive sphingolipids on apoptosis. In K562 CML cell lines resistant to nilotinib, apoptotic ceramide synthase-1 and Bax genes were found to be downregulated while anti-apoptotic SK1 and GCS genes were overexpressed, and inhibition of these overexpressed genes sensitized the cells against nilotinib treatment [208]. Our studies also showed that when we overexpress GCS in imatinib-sensitive K562 cells, the cells developed resistance against the drug via inhibiting apoptosis. Inhibition of GCS and SK1 triggered apoptosis via leading to ceramide accumulation in imatinib-resistant and imatinib-sensitive K562 CML cell lines [209]. Additionally, when we treated K562 human CML cell lines and HL60 human acute pro-myelocytic leukemia cell lines with resveratrol in combination with ceramide analog, GCS inhibitor, or SK1 inhibitor, apoptotic effects of resveratrol increased synergistically in the cells treated with resveratrol and ceramide combination, while the cells treated with resveratrol and GCS or SK1 combinations resisted to apoptosis as compared to the control group [210, 211]. Furthermore, our studies also showed that GCS and SK1 inhibition in combination with nilotinib or dasatinib treatment synergistically triggers apoptosis in K562 and Meg01 human CML cell lines [212, 213].
Briefly, while ceramide is a powerful apoptotic molecule, glucosylceramide and S1P generated from ceramide by GCS and SK1 activities, respectively, are powerful anti-apoptotic molecules, and therefore, alterations in intracellular levels of these sphingolipids could be a novel approach for cancer therapy.
Summary and perspectives
Apoptosis is highly regulated way of cell death, which is crucial for all higher-level organisms to balance tissues homeostasis and control cell proliferation as well as remove damaged or unnecessary cells. Apoptosis has its own morphological and biochemical properties where caspases play a central role at the end. Here, we have focused on aspects of apoptosis in terms of critical genes and their products together with their role in both intrinsic and extrinsic pathways of apoptosis. In this specific vital process, diverse groups of molecules function compatibly for strict regulation. Any alterations or abnormalities occurring in apoptotic processes contribute to development of human diseases and malignancies especially cancer. Deep understanding of apoptotic signaling mechanisms, individual players, and genes involved in apoptosis have provided a great opportunity to develop novel agents that make apoptosis deficient cells sensitive to apoptosis.
References
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2013;35:495–516.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–95.
Horvitz HR. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999;59:1701–6.
Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;10:1456–62.
Krauss G. Biochemistry of signal transduction and regulation. Ed:VCH Wiley, 3rd Edition. 2003;511–531.
Power C, Fanning N, Redmond HP. Cellular apoptosis and organ injury in sepsis: a review. Shoch. 2002;18:197–211.
Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–8.
Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol. 2011;11:1847–57.
Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:255–63.
Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15.
Liu X, Li P, Widlak P, et al. The 40-kDa subunit of DNA fragmentation factor induces DNA fragmentation and chromatin condensation during apoptosis. Proc Natl Acad Sci U S A. 1998;95:8461–6.
Grimsley C, Ravichandran KS. Cues for apoptotic cell engulfment: eat-me, don’t-eat-me and come-get-me signals. Trends Cell Biol. 2003;13:648–56.
Mashima T, Naito M, Noguchi K, Miller DK, Nicholson DW, Tsuruo T. Actin cleavage by CPP-32/apopain during the development of apoptosis. Oncogene. 1997;14:1007–12.
Ziegler U, Groscurth P. Morphological features of cell death. Physiology. 2004;10:124–8.
Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–33.
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6.
Hengartnere MO. The biochemistry of apoptosis. Nature. 2000;407:770–6.
Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55:178–94.
Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–74.
Du C, Fang M, Li Y, Li L, Wang X. SMAC, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42.
Chinnaiyan AM. The apoptosome: heart and soul of the cell death machine. Neoplasia. 1999;1:5–15.
Hill MM, Adrain C, Duriez PJ, Creagh EM, Martin SJ. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. Embo J. 2004;23:2134–45.
Susin SA, Daugas E, Ravagnan L, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–80.
Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56.
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501.
Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–50.
Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–6.
Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–9.
Hitoshi Y, Lorens J, Kitada SI, et al. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity. 1998;8:461–71.
Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–8.
Kuranaga E. Beyond apoptosis: caspase regulatory mechanisms and functions in vivo. Genes Cells. 2012;17:83–97.
Yuan J, Shaham S, Ledoux S, Ellis HM, The HHR. The C. elegans cell death gene Ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–52.
Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–29.
Shi Y. Mechanisms of caspase inhibition and activation during apoptosis. Mol Cell. 2002;9:459–70.
Chowdhury I, Tharakan B, Bhat GK. Caspases—an update. Comp Biochem Physiol. 2008;151:10–27.
Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu Rev Cell Dev Biol. 2005;21:35–56.
Los M, Stroh C, Janicke RU, Schulze-Osthoff K. Caspases: more than just killers? Trends Immunol. 2001;22:31–4.
Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–74.
Los M, van de Craen M, Penning CL, et al. Requirement of an ICE/CED-3 protease for Fas/APO-1-1 mediated apoptosis. Nature. 1995;37:81–3.
Fantuzzi G, Puren AJ, Harding MW, Livingston DJ, Dinarello CA. Interleukin-18 regulation of interferon gamma production and cell proliferation as shown in interleukin-1beta-converting enzyme (caspase-1)-deficient mice. Blood. 1998;91:2118–25.
Vakifahmetoglu-Norberg H, Zhivotovsky B. The unpredictable caspase-2: what can it do? Trends Cell Biol. 2010;20:150–9.
Paroni G, Henderson C, Schneider C, Brancolini C. Caspase-2-induced apoptosis is dependent on caspase-9, but its processing during UV- or tumor necrosis factor-dependent cell death requires caspase-3. J Biol Chem. 2001;276:21907–15.
Van de Craen M, Declercq W. Van den brande I, Fiers W, Vandenabeele P. The proteolytic procaspase activation network: an in vitro analysis. Cell Death Differ. 1999;6:1117–24.
Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–6.
Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–67.
Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104.
Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320–6.
Lakhani SA, Masud A, Kuida K, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–51.
Lamkanfi M, Kanneganti TD. Caspase-7: a protease involved in apoptosis and inflammation. Int J Biochem Cell Biol. 2010;42:21–4.
Ghavami S, Eshraghi M, Kadkhoda K, et al. Role of BNIP3 in TNF-induced cell death—TNF upregulates BNIP3 expression. Biochim Biophys Acta. 1793;2009:546–60.
Carrington PE, Sandu C, Wei Y, et al. The structure of FADD and its mode of interaction with procaspase-8. Mol Cell. 2006;22:599–610.
Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510.
Micheau O, Thome M, Schneider P, et al. Gr ̈utter MG. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–71.
Irmler M, Thome M, Hahne M, et al. Inhibition of death receptors signals by cellular FLIP. Nature. 1997;388:190–5.
Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GC. Activation of caspases-8 and -10 by FLIP (L). Biochem J. 2004;382:651–7.
Pop C, Oberst A, Drag M, et al. FLIP (L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–57.
Chang DW, Xing Z, Pan Y, et al. c-FLIP8(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–14.
Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13:3179–84.
Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999;399:549–57.
Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol. 1993;67:2168–74.
Kaiser WJ, Vucic D, Miller LK. The Drosophila inhibitor of apoptosis D-IAP1 suppresses cell death induced by the caspase drICE. FEBS Lett. 1998;440:243–8.
Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–60.
Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–22.
Banks DP, Plescia J, Altieri DC, et al. Survivin does not inhibit caspase-3 activity. Blood. 2000;96:4002–3.
Saleem M, Qadir MI, Perveen N, et al. Inhibitors of apoptotic proteins: new targets for anticancer therapy. Chem Biol Drug Des. 2013;82:243–51.
Sun C, Cai M, Gunasekera AH, et al. NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature. 1999;401:818–21.
Sun CH, Cai ML, Meadows RP, et al. NMR structure and mutagenesis of the third Bir domain of the inhibitor of apoptosis protein XIAP. J Biol Chem. 2000;275:33777–81.
Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–97.
Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2000;3:401–10.
Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Structural and biochemical basis of apoptotic activation by SMAC/DIABLO. Nature. 2000;406:855–62.
Srinivasula SM, Hegde R, Saleh A, et al. A conserved XIAP- interaction motif in caspase-9 and SMAC/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–6.
Yoo NJ, Kim HS, Kim SY, et al. Immunohistochemical analysis of SMAC/DIABLO expression in human carcinomas and sarcomas. APMIS. 2003;111:382–8.
Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis [IAP] irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17:1487–96.
Liston P, Fong WG, Kelly NL, et al. Identification of XAF1 as an antagonist of XIAP anti-caspase activity. Nat Cell Biol. 2001;3:28–133.
Ma TL, Ni PH, Zhong J, Tan JH, Qiao MM, Jiang SH. Low expression of XIAP- associated factor 1 in human colorectal cancers. Chin J Dig Dis. 2005;6:10–4.
Gross A, Mcdonnell JM, Korsmeyer SJ. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–911.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the Bcl-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;5:49–63.
Pepper C, Bently P. The role of the Bcl-2 family in the modulation of apoptosis. Symp Soc Exp Biol. 2000;52:43–53.
Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9.
Frenzel A, Grespi F, Chmelewskij W, Villunger A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis. 2009;14:584–96.
Camisasca DR, Honorato J, Bernardo V, et al. Expression of Bcl-2 family proteins and associated clinicopathologic factors predict survival outcome in patients with oral squamous cell carcinoma. Oral Oncol. 2009;45:225–33.
Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:126–1132.
Lessene G, Czabotar PE, Colman PM. Bcl-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000.
Hwang JJ, Kuruvilla J, Mendelson D, et al. Phase I dose finding studies of obatoclax (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced solid tumors or lymphoma. Clin Cancer Res. 2010;16:4038–45.
Anderson MA, Huang D, Roberts A. Targeting Bcl2 for the treatment of lymphoid malignancies. Semin Hematol. 2014;51:219–27.
Mahmood Z, Shukla Y. Death receptors: targets for cancer therapy. Exp Cell Res. 2010;316:887–99.
Bhardwaj A, Aggarwal BB. Receptor-mediated choreography of life and death. J Clin Immunol. 2003;23:317–32.
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Sciences. 1998;281:1305–8.
Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–65.
Naismith JH, Sprang SR. Modularity in the TNF receptor family. Trends Biochem Sci. 1998;23:74–9.
Fulda S, Debatin KM. Exploiting death receptor signaling pathways for tumor therapy. Biochim Biophys Acta. 1705;2004:27–41.
Bremer E. Targeting of the tumor necrosis factor receptor superfamily for cancer immunotherapy. ISRN Oncol. 2013;2013:371854.
Walczak H, Krammer PH. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res. 2000;256:58–66.
Behrmann I, Walczak H, Krammer PH. Structure of the human APO-1 gene. Eur J Immunol. 1994;24:3057–62.
Tauzin S, Debure L, Moreau JF, Legembre P. CD95-mediated cell signaling in cancer: mutations and posttranslational modulations. Cell Mol Life Sci. 2012;69:1261–77.
Scholl V, Stefanoff CG, Hassan R, Spector N, Renault IZ. Mutations within the 5′ region of FAS/CD95 gene in nodal diffuse large B-cell lymphoma. Leuk Lymphoma. 2007;48:957–63.
Ivanov VN, Ronai Z, Hei TK. Opposite roles of FAP-1 and dynamin in the regulation of Fas (CD95) translocation to the cell surface and susceptibility to Fas ligand-mediated apoptosis. J Biol Chem. 2006;281:1840–52.
Tourneur L, Mistou S, Michiels FM, et al. Loss of FADD protein expression results in a biased Fas-signaling pathway and correlates with the development of tumoral status in thyroid follicular cells. Oncogene. 2003;22:2795–280.
Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene. 2001;20:5865–77.
Yang T, Shi R, Chang L, et al. Huachansu suppresses human bladder cancer cell growth through the Fas/Fasl and TNF-alpha/TNFR1 pathway in vitro and in vivo. J Exp Clin Cancer Res. 2015;34:1–10.
Zhong W, Qin S, Zhu B, et al. Oxysterol-binding protein-related protein 8 [ORP8] increases sensitivity of hepatocellular carcinoma cells to Fas-mediated apoptosis. J Biol Chem. 2015;290:8876–87.
Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–90.
Mahalingam D, Szegezdi E, Keane M, de Jong S, Samali A. TRAIL receptor signalling and modulation: are we on the right TRAIL? Cancer Treat Rev. 2009;35:280–8.
Wu GS. TRAIL as a target in anti-cancer therapy. Cancer Lett. 2009;285:1–5.
O’Leary L, van der Sloot AM, Reis CR, et al. Decoy receptors block TRAIL sensitivity at a supracellular level: the role of stromal cells in controlling tumour TRAIL sensitivity. Oncogene. 2015. doi:10.1038/onc.2015.180.
Woo JK, Kang JH, Jang YS, et al. Evaluation of preventive and therapeutic activity of novel non-steroidal anti-inflammatory drug, CG100649, in colon cancer: increased expression of TNF-related apoptosis-inducing ligand receptors enhance the apoptotic response to combination treatment with TRAIL. Oncol Rep. 2015;3:1947–55.
Emery JG, McDonnell P, Burke MB, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–7.
Weichhaus M, Chung ST, Connelly L. Osteoprotegerin in breast cancer: beyond bone remodeling. Mol Cancer. 2015;14:117.
Lane D, Matte I, Rancourt C, Piché A. Osteoprotegerin [OPG] protects ovarian cancer cells from TRAIL-induced apoptosis but does not contribute to malignant ascites-mediated attenuation of TRAIL-induced apoptosis. J Ovarian Res. 2012;5:34.
Merino D, Lalaoui N, Morizot A, Solary E, Micheau O. TRAIL in cancer therapy: present and future challenges. Expert Opin Ther Targets. 2007;11:1299–314.
Gong B, Almasan A. Genomic organization and transcriptional regulation of human APO2/TRAIL gene. Biochem Biophys Res Commun. 2000;278:747–52.
Krieg A, Krieg T, Wenzel M, et al. TRAIL-beta and TRAIL-gamma: two novel splice variants of the human TNF-related apoptosis- inducing ligand (TRAIL) without apoptotic potential. Br J Cancer. 2003;88:918–27.
Pal R, Gochhait S, Chattopadhyay S, et al. Functional implication of TRAIL-716 C/T promoter polymorphism on its in vitro and in vivo expression and the susceptibility to sporadic breast tumor. Breast Cancer Res Treat. 2012;126:333–43.
Bos PD, Zhang XHF, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005–9.
Allen JE, El-Deiry WS. Regulation of the human TRAIL gene. Cancer Biol Ther. 2012;13:1143–51.
Lim B, Allen JE, Prabhu VV, Talekar MK, Finnberg NK, El-Deiry WS. Targeting TRAIL in the treatment of cancer: new developments. Expert Opin Ther Targets. 2015;25:1–15.
Falvo JV, Tsytsykova AV, Goldfeld AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun. 2010;11:27–60.
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65.
Sedger LM, McDermott MF. TNF and TNF-receptors: from mediators of cell death and inflammation to therapeutic giants—past, present and future. Cytokine Growth Factor Rev. 2014;25:453–72.
Wachter T, Sprick M, Hausmann D, et al. cFLIPL inhibits tumor necrosis factor-related apoptosis-inducing ligand-mediated NF-kappaB activation at the death-inducing signaling complex in human keratinocytes. J Biol Chem. 2004;279:52824–34.
Ebach DR, Riehl TE, Stenson WF. Opposing effects of tumor necrosis factor receptor 1 and 2 in sepsis due to cecal ligation and puncture. Shock. 2005;23:311–8.
Yun HM, Park KR, Kim EC, Han SB, Yoon do Y, Hong JT. IL-32α suppresses colorectal cancer development via TNFR1-mediated death signaling. Oncotarget. 2015;6:9061–72.
Yu S, Hou D, Chen P, et al. Adenosine induces apoptosis through TNFR1/RIPK1/P38 axis in colon cancer cells. Biochem Biophys Res Commun. 2015;460:759–65.
Bake V, Roesler S, Eckhardt I, Belz K, Fulda S. Synergistic interaction of SMAC mimetic and IFNα to trigger apoptosis in acute myeloid leukemia cells. Cancer Lett. 2014;355:224–31.
Tao YF, Lu J, Du XJ, et al. Survivin selective inhibitor YM155 induce apoptosis in SK-NEP-1 Wilms tumor cells. BMC Cancer. 2012;26(12):619.
Ruddle NH. Lymphotoxin and TNF: how it all began—a tribute to the travelers. Cytokine Growth Factor Rev. 2014;25:83–9.
Chaturvedi MM, LaPushin R, Aggarwal BB. Tumor necrosis factor and lymphotoxin. Qualitative and quantitative differences in the mediation of early and late cellular response. J Biol Chem. 1994;269:14575–83.
Etemadi N, Holien JK, Chau D, et al. Lymphotoxin α induces apoptosis, necroptosis and inflammatory signals with the same potency as tumour necrosis factor. FEBS J. 2013;280:5283–97.
de Oliveira JG, Rossi AF, Nizato DM, et al. Influence of functional polymorphisms in TNF-α, IL-8, and IL-10 cytokine genes on mRNA expression levels and risk of gastric cancer. Tumour Biol. 2015 (Epub ahead of print).
Kang YJ, Kim WJ, Bae HU, et al. Involvement of TL1A and DR3 in induction of proinflammatory cytokines and matrix metalloproteinase-9 in atherogenesis. Cytokine. 2005;29:229–35.
Lee SY, Debnath T, Kim SK, Lim BO. Anti-cancer effect and apoptosis induction of cordycepin through DR3 pathway in the human colonic cancer cell HT-29. Food Chem Toxicol. 2013;60:439–47.
Oh SB, Hwang CJ, Song SY, et al. Anti-cancer effect of tectochrysin in NSCLC cells through overexpression of death receptor and inactivation of STAT3. Cancer Lett. 2014;353:95–103.
Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58.
Surget S, Khoury MP, Bourdon J. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57–68.
Mollereau B, Ma D. The p53 control of apoptosis and proliferation: lessons from Drosophila. Apoptosis. 2014;19:1421–9.
Pflaum J, Schlosser S, Müller M. p53 family and cellular stress responses in cancer. Front Oncol. 2014;4:285.
Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935.
Chi SW. Structural insights into the transcription-independent apoptotic pathway of p53. BMB Rep. 2014;47:167–72.
Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis—the p53 network. J Cell Sci. 2003;116:4077–85.
Perry ME. The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb Perspect Biol. 2010;2:a000968.
Riley MF, You MJ, Multani AS, Lozano G. Mdm2 overexpression and p73 loss exacerbate genomic instability and dampen apoptosis, resulting in B-cell lymphoma. Oncogene. 2015. doi:10.1038/onc.2015.88(Epubaheadofprint).
Jansson MD, Damas ND, Lees M, Jacobsen A, Lund AH. miR-339-5p regulates the p53 tumor-suppressor pathway by targeting MDM2. Oncogene. 2014;34:1908–18.
Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene Suppl. 1998;1:S71–83.
Hikisz P, Kiliańska ZM. PUMA, a critical mediator of cell death—one decade on from its discovery. Cell Mol Biol Lett. 2012;17:646–69.
Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–57.
MacLachlan TK, El-Deiry WS. Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc Natl Acad Sci U S A. 2002;99:9492–7.
Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90.
Ha JH, Shin JS, Yoon MK, et al. Dual-site interactions of p53 protein transactivation domain with anti-apoptotic Bcl-2 family proteins reveal a highly convergent mechanism of divergent p53 pathways. J Biol Chem. 2013;288:7387–98.
Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4.
Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1complex. Nat Cell Biol. 2004;6:443–50.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinicaluse. Cold Spring Harb Perspect Biol. 2010;2:a001008.
Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–13.
Saleem S, Abbasi ZA, Hameed A, Qureshi NR, Khan MA, Azhar A. Novel p53 codon 240 Ser > Thr coding region mutation in the patients of oral squamous cell carcinoma (OSCC). Tumour Biol. 2014;35:7945–50.
Trbusek M, Smardova J, Malcikova J, et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol. 2011;29:2703–8.
Wang S, Zhou M, Ouyang J, Geng Z, Wang Z. Tetraarsenictetrasulfide and arsenic trioxide exert synergistic effects on induction of apoptosis and differentiation in acute promyelocytic leukemia cells. PLoS One. 2015;10:e0130343.
Gu ZT, Li L, Wu F, et al. Heat stress induced apoptosis is triggered by transcription-independent p53, Ca[2+] dyshomeostasis and the subsequent Bax mitochondrial translocation. Sci Rep. 2015;5:11497.
Sosin AM, Burger AM, Siddiqi A, Abrams J, Mohammad RM, Al-Katib AM. HDM2 antagonist MI-219 [spiro-oxindole], but not Nutlin-3 [cis-imidazoline], regulates p53 through enhanced HDM2 autoubiquitination and degradation in human malignant B-cell lymphomas. J Hematol Oncol. 2012;5:57.
Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A. 2008;105:10360–5.
Vermeij R, Leffers N, van der Burg SH, Melief CJ, Daemen T, Nijman HW. Immunological and clinical effects of vaccines targeting p53-overexpressing malignancies. J Biomed Biotechnol. 2011;2011:702146.
Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH. MicroRNA regulation of core apoptosis pathways in cancer. Eur J Cancer. 2011;47:163–74.
Chen Y, Fu LL, Wen X, et al. Oncogenic and tumor suppressive roles of microRNAs in apoptosis and autophagy. Apoptosis. 2014;19:1177–89.
Acunzo M, Visone R, Romano G, et al. Mir-130a targets MET and induces trail-sensitivity in NSCLC by downregulating mir-221 and 222. Oncogene. 2012;31:634–42.
Hao J, Zhang C, Zhang A, et al. miR-221/222 is the regulator of Cx43 expression in human glioblastoma cells. Oncol Rep. 2012;27:1504–10.
Wang P, Zhuang L, Zhang J, et al. The serum miR-21 level serves as a predictor for the chemosensitivity of advanced pancreatic cancer, and miR-21 expression confers chemoresistance by targeting FasL. Mol Oncol. 2013;7:334–45.
Qin W, Shi Y, Zhao B, et al. miR-24 regulates apoptosis by targeting the open reading frame (ORF) region of FAF1 in cancer cells. PLoS ONE. 2010;5:e9429.
Satzger I, Mattern A, Kuettler U, et al. microRNA-21 is upregulated in malignant melanoma and influences apoptosis of melanocytic cells. Exp Dermatol. 2012;21:509–14.
Eto K, Iwatsuki M, Watanabe M, et al. The microRNA-21/PTEN pathway regulates the sensitivity of HER2-positive gastric cancer cells to trastuzumab. Ann Surg Oncol. 2013;21:343–50.
Schickel R, Park SM, Murmann AE, Peter ME. miR-200c regulates induction of apoptosis through CD95 by targeting FAP-1. Mol Cell. 2010;38:908–15.
Li JH, Xiao X, Zhang YN, et al. MicroRNA miR-886-5p inhibits apoptosis by down-regulating Bax expression in human cervical carcinoma cells. Gynecol Oncol. 2011;120:145–51.
Zhou M, Liu Z, Zhao Y, et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem. 2010;285:21496–507.
Zhang H, Zuo Z, Lu X, Wang L, Wang H, Zhu Z. MiR-25 regulates apoptosis by targeting Bim in human ovarian cancer. Oncol Rep. 2012;27:594–8.
Gocek E, Wang X, Liu X, Liu CG, Studzinski GP. MicroRNA-32 upregulation by 1,25-dihydroxyvitamin D3 in human myeloid leukemia cells leads to Bim targeting and inhibition of AraC-induced apoptosis. Cancer Res. 2011;71:6230–9.
Veronese A, Lupini L, Consiglio J, et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010;70:3140–9.
Tanaka N, Toyooka S, Soh J, et al. Downregulation of microRNA-34 induces cell proliferation and invasion of human mesothelial cells. Oncol Rep. 2013;29:2169–74.
Shen J, Wan R, Hu G, et al. miR-15b and miR-16 induce the apoptosis of rat activated pancreatic stellate cells by targeting Bcl-2 in vitro. Pancreatology. 2012;12:91–9.
Xu J, Liao X, Wong C. Downregulations of B-cell lymphoma 2 and myeloid cell leukemia sequence 1 by microRNA 153 induce apoptosis in a glioblastoma cell line DBTRG-05MG. Int J Cancer. 2010;126:1029–35.
Nakano H, Miyazawa T, Kinoshita K, Yamada Y, Yoshida T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-X(L) in colorectal cancer cells. Int J Cancer. 2010;127:1072–80.
Shang J, Yang F, Wang Y, et al. Sun S MicroRNA-23a antisense enhances 5-fluorouracil chemosensitivity through APAF-1/caspase-9 apoptotic pathway in colorectal cancer cells. J Cell Biochem. 2014;115:772–84.
Walker JC, Harland RM. MicroRNA-24a is required to repress apoptosis in the developing neural retina. Genes Dev. 2009;23:1046–51.
Wu JH, Yao YL, Gu T, et al. MiR-421 regulates apoptosis of BGC-823 gastric cancer cells by targeting caspase-3. Asian Pac J Cancer Prev. 2014;15:5463–8.
Hudson RS, Yi M, Esposito D, et al. Microrna-106b-25 cluster expression is associated with early disease recurrence and targets caspase-7 and focal adhesion in human prostate cancer. Oncogene. 2013;32:4139–47.
Floyd DH, Zhang Y, Dey BK, et al. Novel anti-apoptotic microRNAs 582–5p and 363 promote human glioblastoma stem cell survival via direct inhibition of caspase 3, caspase 9, and Bim. PLoS One. 2014;9:e96239.
Tsang WP, Kwok TT. Let-7a microRNA suppresses therapeutics-induced cancer cell death by targeting caspase-3. Apoptosis. 2008;13:1215–22.
Zhang J, Du Y, Wu C, et al. Curcumin promotes apoptosis in human lung adenocarcinoma cells through miR-186 signaling pathway. Oncol Rep. 2010;24:1217–23.
Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–50.
Barth BM, Cabot MC, Kester M. Ceramide-based therapeutics for the treatment of cancer. Anti Cancer Agents Med Chem. 2011;11:911–9.
Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism—a strategy for overcoming drug resistance. J Natl Cancer Inst. 2001;93:347–57.
Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem. 2002;277:26796–803.
Siskind LJ, Feinstein L, Yu TX, et al. Anti-apoptotic Bcl-2 family proteins disassemble ceramide channels. J Biol Chem. 2008;283:6622–30.
von Haefen C, Wieder T, Gillissen B, et al. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–19.
Dumitru CA, Sandalcioglu IE, Wagner M, Weller M, Gulbins E. Lysosomal ceramide mediates gemcitabine-induced death of glioma cells. J Mol Med. 2009;87:1123–32.
Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer: pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–16.
Liu F, Verin AD, Wang P, et al. Differential regulation of sphingosine-1-phosphate- and VEGF-induced endothelial cell chemotaxis. Involvement of G(ialpha2)-linked Rho kinase activity. Am J Respir Cell Mol Biol. 2001;24:711–9.
Radin NS. The development of aggressive cancer: a possible role for sphingolipids. Cancer Investig. 2002;20:779–86.
Pchejetski D, Golzio M, Bonhoure E, et al. Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res. 2005;65:11667–75.
Beckham TH, Lu P, Jones EE, et al. LCL124, a cationic analog of ceramide, selectively induces pancreatic cancer cell death by accumulating in mitochondria. J Pharmacol Exp Ther. 2013;344(1):167–78.
Jiang Y, DiVittore NA, Kaiser JM, et al. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol Ther. 2011;12(7):574–85.
Sorli SC, Colié S, Albinet V, et al. The nonlysosomal β-glucosidase GBA2 promotes endoplasmic reticulum stress and impairs tumorigenicity of human melanoma cells. FASEB J. 2013;27(2):489–98.
Stover TC, Sharma A, Robertson GP, Kester M. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res. 2005;11(9):3465–74.
Beljanski V, Lewis CS, Smith CD. Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in hepatocellular carcinoma xenografts. Cancer Biol Ther. 2011;11(5):524–34.
Adan-Gokbulut A, Kartal-Yandim M, Iskender G, Baran Y. Novel agents targeting bioactive sphingolipids for the treatment of cancer. Curr Med Chem. 2013;20(1):108–22.
Coward J, Ambrosini G, Musi E, Truman JP, Haimovitz-Friedman A, Allegood JC. Safingol (L-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy. 2009;5:184–93.
Pyne S, Bittman R, Pyne NJ. Sphingosine kinase inhibitors and cancer: seeking the golden sword of Hercules. Cancer Res. 2011;71(21):6576–82.
Taouji S, Higa A, Delom F, et al. Phosphorylation of serine palmitoyltransferase long chain-1 (SPTLC1) on tyrosine 164 inhibits its activity and promotes cell survival. J Biol Chem. 2013;288(24):17190–201.
Huang WC, Tsai CC, Chen CL, et al. Glucosylceramide synthase inhibitor PDMP sensitizes chronic myeloid leukemia T315I mutant to Bcr-Abl inhibitor and cooperatively induces glycogen synthase kinase-3-regulated apoptosis. FASEB J. 2011;25(10):3661–73.
Nica AF, Tsao CC, Watt JC, et al. Ceramide promotes apoptosis in chronic myelogenous leukemia-derived K562 cells by a mechanism involving caspase-8 and JNK. Cell Cycle. 2008;7(21):3362–70.
Camgoz A, Gencer EB, Ural AU, Baran Y. Mechanisms responsible for nilotinib resistance in human chronic myeloid leukemia cells and reversal of resistance. Leukemia Lymphoma. 2013;54:1279–87.
Baran Y, Bielawski J, Gunduz U, Ogretmen B. Targeting glucosylceramide synthase sensitizes imatinib-resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. J Cancer Res Clin Oncol. 2011;137:1535–44.
Kartal M, Saydam G, Sahin F, Baran Y. Resveratrol triggers apoptosis through regulating ceramide metabolizing genes in human K562 chronic myeloid leukemia cells. Nutr Cancer. 2011;63(4):637–44.
Cakir Z, Saydam G, Sahin F, Baran Y. The roles of bioactive sphingolipids in resveratrol-induced apoptosis in HL60: acute myeloid leukemia cells. J Cancer Res Clin Oncol. 2011;137(2):279–86.
Camgoz A, Gencer EB, Ural AU, Avcu F, Baran Y. Roles of ceramide synthase and ceramide clearence genes in nilotinib-induced cell death in chronic myeloid leukemia cells. Leukemia Lymphoma. 2011;52:1574–84.
Gencer EB, Ural AU, Avcu F, Baran Y. A novel mechanism of dasatinib-induced apoptosis in chronic myeloid leukemia; ceramide synthase and ceramide clearance genes. Ann Hematol. 2011;90:1265–75.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None
Rights and permissions
About this article

Cite this article
Kiraz, Y., Adan, A., Kartal Yandim, M. et al. Major apoptotic mechanisms and genes involved in apoptosis. Tumor Biol. 37, 8471–8486 (2016). https://doi.org/10.1007/s13277-016-5035-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5035-9