
Abstract
During the past decade, a heightened understanding of metabolic pathways in cancer has significantly increased. It is recognized that many tumor cells are genetically programmed and have involved an abnormal metabolic state. Interestingly, this increased metabolic autonomy generates dependence on various nutrients such as glucose and glutamine. Both of these components participate in various facets of metabolic activity that allow for energy production, synthesis of biomass, antioxidant defense, and the regulation of cell signaling. Here, we outline the emerging data on glutamine metabolism and address the molecular mechanisms underlying glutamine-induced cell survival. We also discuss novel therapeutic strategies to exploit glutamine addiction of certain cancer cell lines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Early observations put forward the idea that mouse tumor cells were different from differentiated tissues in that they metabolize glucose by reprogramming their fermentative energy metabolism pathway even when oxygen is abundant [1, 2]. Although this term is coined “the Warburg effect,” it is a misinterpretation of damage in mitochondrial respiration instead of damage to the regulation of glycolysis. It has governed basic research and clinical implications over decades, leading, for instance, to the development of the clinical procedure known as 18fluoro-2-deoxyglucose imaging by positron emission tomography (PET), which has been used as a diagnostic test [3, 4]. The metabolic state of tumor cells have recently re-emerged and are more complicated than previously thought. The metabolic state is primarily based on nutrients available to the tumors which have rewired their metabolic pathways, thereby contributing to this metabolic autonomy [5–8]. The discoveries of oncogenes and tumor suppressors that regulate nutrient uptake and its utilization have provided insights that nutrients per se play a pivotal role in cell growth and proliferation through a wide array of cell signaling pathways [9–15]. It is important to note that several lines of evidence suggest that glutamine is an essential requirement to support anabolic metabolism in tumor cells. This sheds new light on the importance of this nutrient to cell growth, proliferation, malignancy, and stress resistance [16]. For instance, extracellular glutamine can donate carbon and nitrogen to supply anabolic pathways and energy production. It has the ability to replenish TCA cycle intermediaries (anaplerosis) and promote synthesis of nucleotides, proteins, and lipids [17]. Although glutamine contributes to anaplerosis in many tumor cells, in glioblastomas, it has been recently described as a cataplerotic mechanism, where glutamine-derived glutamate is secreted and does not enter the TCA cycle. This mechanism relies on glutamine synthetase (GS) activity, the enzyme that catalyzes, using ATP, the synthesis of glutamine from glutamate and ammonia (NH4+), leading to nucleotide biosynthesis and support glioblastoma growth [18]. Moreover, glutamine might be robustly correlated, rather than glucose, to maintain cancer invasion in ovarian tumor growth and in overall patient survival [19]. Conversely, an elegant observation has shown that other nutrients such as aspartate and asparagine can prevent cancer cell death during glutamine withdrawal [20]. Together, these findings are paving the way to understand glutamine metabolism and to exploit the metabolic nutrient state of susceptible populations of tumor cells for cancer therapy.
In line with this notion, a substantial understanding of how and which nutrients are important for proliferating tumor cells, why some metabolic pathways are more important than others to accompany tumorigenesis, and how the intricate glutamine metabolism may be exploited for new therapeutic approaches are also highlighted in this review.
Glutamine metabolism
Previous studies in cell culture and mouse models have suggested the importance of glutamine for proliferating tumor cells [21, 22]. In various physiological conditions, glutamine is considered a nonessential amino acid (NEAA). In plasma, it is found at a concentration of 0.6–0.9 mmol/L (most abundant), and many tissues can synthesize this amino acid [23]. However, in pathophysiology, glutamine might turn essential, reflecting the avid glutamine consumption by certain cancer cell lines [24].
Glutamine metabolism begins with the uptake of glutamine, which can be performed by four different families of amino acid transporters. In fact, differential expression of each transporter relies on cell type and the tissue that requires glutamine. Interestingly, early observations have identified as overexpressed the neutral amino acid transporter (ASCT2) in human colorectal adenocarcinoma [25]. The main member of the ASC family of transporters, also known as solute linked carrier family 1 member A5 (SLC1A5), acts as a high affinity transporter of glutamine, and it has been shown to be upregulated in a diverse panel of cultured human cancer cell lines [26–28]. Further analysis showed that the inhibition of SLC1A5 decreased uptake of L-glutamine, inhibited the mammalian target of rapamycin (mTOR), triggered autophagy, and cell death [29, 30]. After its uptake, glutamine catabolism begins with an initial deamination converting glutamine to glutamate, in a reaction catalyzed by a phosphate-dependent glutaminase (GLS1 and GLS2). This reaction also generates NH4+, a metabolic by-product that stimulates autophagy (see “Depot for ammonia and autophagy”) [31]. Recently, GLS1 has been described as an enzyme induced by the oncogene c-Myc (hereafter cited as Myc), which enhances GLS1 expression and glutamine catabolism to support cell survival and proliferation [11]. The role of GLSs and NH4+ is addressed below (see “Glutamine: paradoxical roles in cancer metabolism”). Moreover, glutamine can also supply amino groups for the hexosamine biosynthetic pathway (HBP) [32]. The enzyme, fructose-6-phosphate amidotransferase receives glutamine’s amino group to generate fructose-6-phosphate, and then glucosamine-6-phosphate, a key metabolite for posttranslational modification, including N-linked and O-linked glycosylation reactions [33]. In addition, L-glutamine serves in exchange with the import of other amino acids such as arginine, cystine, leucine, and others [29, 34, 35]. Finally, glutamine can directly donate carbon and nitrogen for the synthesis of nucleotides (e.g., purine and pyrimidines). Aspartate and glycine can participate in the de novo synthesis of nucleotides, as glutamine supports their production [33].
Glutamate can remain in the cytoplasm to be used for glutathione synthesis and favor the intracellular redox homeostasis. When the cytoplasmic environment is rich in glutamate, cysteine, glycine, and ATP, the enzyme glutathione synthetase generates reduced glutathione (GSH). A study using a panel of human cancer cell lines has shown that the intracellular redox balance between reduced and oxidized glutathione (GSSG) represents one of the most important antioxidant systems in cells [13]. Glutamate serves as a source of amino groups for NEAA production, particularly aspartate and alanine. This set of reactions transfers amino groups between glutamate and α-ketoacid which is used to synthesize proteins [16]. Moreover, glutamate can enter into mitochondria and be oxidized to α-ketoglutarate (α-KG), reducing NADP+ to NADPH or NAD+ to NADH and produces a second molecule of NH4+. This reaction is catalyzed by the glutamate dehydrogenase (GDH) which also plays a pivotal role in cancer survival [36]. GDH is the unique enzyme that can use NADP+ or NAD+ as cofactor. During mitochondrial metabolism, α-KG can support TCA cycle intermediaries through oxidation of succinly-CoA and by reducing NAD+ to NADH to maintain mitochondrial integrity and activity. In general, mitochondrial glutamine catabolism donates carbon skeletons to replenish TCA cycle intermediaries, as malate is converted to pyruvate, and finally transformed to lactate or alanine (glutaminolysis). Yet, the production of malate plus the activity of a particular aminotransferase enzyme (TA), known as aspartate-aminotransferase, represents a central cellular redox balance route between mitochondria and the cytoplasm. The malate-aspartate shuttle is able to transfer electrons to mitochondrial complex I, generating ATP, and resupplying NAD+ that drives glycolysis. Moreover, oxalacetate (OAA), another TCA cycle intermediary, in the presence of acetyl-CoA, can generate citrate, providing de novo fatty acid biosynthesis [17].
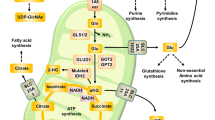
Intriguingly, it has been described that glutamine acts as a compensatory metabolic route to maintain cancer progression. For instance, recent findings showed an unknown role of glutamine in a panel of human tumor cell lines with defective mitochondria or under a hypoxia condition. In particular, these cell lines produce citrate by reductively carboxylating α-KG via NADPH-dependent isocitrate dehydrogenase (IDH), supporting lipogenesis [37, 38]. In agreement with this study, some somatic mutations in the IDH-1 and IDH-2 genes have been found in gliomas and acute myelogenous leukemia (AML). The mutations permit the ability to convert α-KG to the metabolite 2-hydroxyglutarate (2-HG) [39]. The accumulation of this oncometabolite (a small molecule of normal metabolism whose accumulation causes metabolic dysregulation) has been linked with this metabolic switch and an altered epigenetic state (e.g., histone and DNA dimethylation) which is advantageous to promote cellular transformation [40]. Moreover, in certain K-ras-mutant cell lines, macropinocytosis can support uptake of extracellular protein to provide amino acid supplies (e.g., glutamine) for cell growth [41]. Conversely, some evidence suggests that the role of glutamine in anaplerosis is not the direct mechanism to avoid apoptosis, and that enhanced oxidation of branched chain amino acid (BCAA), as valine, leucine, and isoleucine, can also fuel tumor cells’ bioenergetic demand [20]. This metabolic state of malignant cells challenges the current model and opens up interest to get a deeper understanding of this profound and intricate interplay between glutamine and cancer metabolic reprogramming. The entire role of glutamine metabolism has been summarized in a schematic diagram, in Fig. 1.

Certain tumor cell lines rely on glutamine utilization to support growth, proliferation, and survival. The import and export of glutamine is performed through surface transporters, like SLC1A5 and SLC7A5, and in the exchange for the uptake of extracellular EAAs, respectively. Moreover, in certain K-ras-mutant cell lines, macropinocytosis can support uptake of extracellular protein to provide amino acids such as glutamine. In the cytoplasm, glutamine serves as primary source of nitrogen, which supports biosynthetic production, including de novo nucleotide and amino acid synthesis, hexosamine biosynthetic pathway (glusosamide), and glutathione synthesis (GSSG/GSH). In mitochondria, glutamine anaplerosis replenishes TCA cycle intermediates as precursors for macromolecule production such as citrate, which is used to fuel lipogenesis. In addition, mitochondrial glutamine can generate energy through ATP production via OXPHOS. During glutaminolysis, both enzymes GLS and GDH produce NH4+, which plays a central role in autophagy and cancer progression. Glucose metabolism represents a source of ATP as well as a source of biosynthetic precursors for tumor growth. EAA essential amino acids, Glu glutamate, Cys cysteine, Lac lactate, xC exchange amino acid transport, MCT4 lactate transporter, α-KG α-keto-glutarate, NEAA nonessential amino acid, Gly glycine, OAA oxalacetate, Pry pyruvate, AcCoA acetyl coenzyme A
Glutamine: paradoxical roles in cancer metabolism
Oncogenic and tumor suppressor signaling pathways
C. Shih et al. were the first to attribute that cancer was a disease of altered genes [42]. However, the study carried out by H. Varmus and J. M. Bishop boosted our knowledge of cancer, leading them to win a Nobel Prize in 1989. The authors showed that oncogenes were not actually viral genes and might arise from genes normally present in cells [43]. As a consequence of this seminal discovery, various oncogenes and tumor suppressor genes have long been described to alter cell physiology, including Myc, Ras, p53, PI3K, Akt, Her-2, PTEN, and others [44].
Traditionally, oncogenes and tumor suppressor genes were first indentified as regulators of the cell cycle and proliferative signaling pathways. They were later found to regulate growth suppression and cell death. Conversely, recent findings have showed that oncogenes and tumor suppressor genes orchestrate metabolic needs of cells while proliferating. These insights brought renewed attention to the study of metabolic pathways involved in tumor growth. Medina and colleagues were the first to discuss the role of glutamine metabolism in cancer [45], but only recently, DeBerardinis et al. have observed that transformed cells rely on glutamine metabolism to support the synthesis of protein and nucleotides, despite aerobic glycolysis. By using a 13CNMR spectroscopy technique to study gliomas, the authors showed the catabolic fate of glutamine, which supports the synthesis of macromolecules and the intracellular redox balance. They showed that glutamine seems to maintain cell survival by providing anaplerotic precursors to maintain TCA cycle intermediates, glutaminolysis, and high levels of NADPH [17]. In this current section, we highlight the versatile role of glutamine and the intricate interplay between this amino acid and the most notorious oncogenes and tumor suppressor gene signaling pathways.
Myc
The first to appreciate that glutamine withdrawal induces Myc-dependent apoptosis were Yuneva et al. They demonstrated that apoptosis precedes TCA cycle intermediary depletion and might be reverted when cells were incubate in the presence of pyruvate and OAA [15]. Mechanistically, Myc regulates glutamine uptake and glutaminolysis due in part by increasing the transcriptional expression of glutamine transporters, GLS1, and lactate dehydrogenase (LDH). Accordingly, despite glucose being present, Myc can induce a metabolic switch leading to glutamine addiction. This is required to maintain anaplerosis and mitochondrial activity. Subsequent experiments showed that Myc-transformed cells, when challenged with an inhibitor of glutamate-dependent transaminases [known as aminooxyacetate (AOA)], activated the intrinsic apoptotic pathway as the mechanism of cell death. Interestingly, when Myc-transformed cells were supplemented with cell-permeable α-KG, these cells increased sensitivity to resist apoptosis [14]. Other studies have shown that Myc regulates glutamine metabolism in human cancer cells by transcriptionally repressing microRNAs: known as miR-23a/b. This results in increased mitochondrial GLS1 expression. The authors found that Myc-induced glutamine catabolism seems to provide energy through mitochondrial activity and to maintain a reducing equivalent avoiding oxidative stress [11].
In fact, some studies have shown unquestionable evidence that targeting glutamine metabolism, like blocking mitochondrial GLS1 activity, could be used to suppress tumor cell growth and transformation [36, 46]. However, recent findings have suggested that even silencing GLS1 could suppress, but not eliminate, glioblastoma cell growth in vitro and in vivo. By using a metabolic flux technique, the authors found a compensatory anaplerotic mechanism pyruvate-carboxlyase-dependent (PC) that provides OAA and pyruvate, resulting in resistance to apoptosis in a glutamine-depleted state [47]. In line with this notion, to investigate how glutamine catabolism can suppress apoptosis, Zhang et al. screened by RNAi-based techniques factors what could protect Myc-transformed cells from apoptosis during glutamine withdrawal. The authors found that knockdown of citrate synthase (CS) resulted in redirection of OAA into aspartate and asparagines biosynthesis, blocking cell death [20].
Remarkably, in neuroblastomas with high N-Myc expression, a particular apoptosis cell death pathway (ATF4-dependent), but p53-independent (during glutamine depletion) was indentified. The authors showed that apoptosis could be prevented by restoring TCA cycle intermediaries such as α-KG and OAA [48]. Nonetheless, other recent findings also showed that breast cancer cells with a Myc signature accumulated 2-HG in the presence of glutamine [49].
Glutamine anaplerosis is transcriptionally regulated by Myc. Surprisingly, it has only recently been proposed that a number of human and murine cells undergoing aerobic glycolysis, and upon glutamine limitation, can upregulate the expression of GS. The mechanism by which Myc regulates GS activity is directly associated with thymine DNA glycosylase (TDG). TDG promotes active demethylation of the GS promoter and its increased expression [50]. Therefore, de novo glutamine synthesis serves as a metabolic route used for cancer cells rather than glutamine anaplerosis through the TCA cycle. Thus, it is reasonable to speculate that in tumor tissue, under glutamine shortage, there is a mechanism that supports de novo glutamine synthesis. In fact, Kung et al. showed that basal-type, but not luminal-type, breast cells rely on glutamine utilization, while luminal cells can also rescue basal cells in coculture during glutamine limitation. There is due to luminal-specific expression of GS, which is directly induced by GATA3, which represses the glutaminase expression [51].
Thus, the complex mechanism by which Myc-induced glutamine catabolism and anabolism has heightened our comprehension of tumor biology of certain cancer cells and may prove to be more useful in cancer therapies.
K-ras
The Ras family of proteins is involved in signal transduction from tyrosine kinase receptor and plays a key role in cell growth and survival [52]. Early evidence has showed that cells harboring constitutive K-ras expression increased glucose uptake and lactate production [53–56]. Subsequent reports have also demonstrated that human colon cancer cell lines and K-ras transformed NIH3T3 fibroblast could also rely upon glutamine metabolism through functional mitochondrial activity [10, 57]. A comprehensive analysis of metabolic reprogramming in K-ras transformed NIH3T3 fibroblast and human breast cell lines (K-ras-mutant) was performed later by Gaglio et al. The authors showed that K-ras transformed fibroblast decoupled glucose and glutamine metabolism leading to an increase utilization of carbon skeleton and amino-nitrogen glutamine-derivate through the TCA cycle or transamination activities to sustain macromolecule biosynthesis [58].
Recently, a noncanonical pathway of glutamine metabolism in human pancreatic ductal adenocarcinoma (PDAC) cells that regulates cellular redox homeostasis has been indentified, and it is required for these tumors to grow. The authors found an unusual route where glutamine is converted into aspartate, and it can be transported to the cytoplasm through the malate-aspartate shuttle. In the cytoplasm, aspartate can be converted into OAA by the activity of aspartate transaminase (GOT1). Subsequently, OAA is converted to malate and pyruvate, which are used by malic enzyme (ME1) to sustain the NADPH/NADP+ ratio, which maintains cellular redox homeostasis [13]. Since the oncogenic K-ras-mutation is present in more than 95 % of PDAC, this oncogene has been considered as a master regulator pathway that uses glutamine metabolism to decrease intracellular ROS levels, contributing significantly to cell proliferation and tumorigenesis [59]. In this regard, it was reasonable to speculate that the tumor microenvironment of PDAC usually contains depleted levels of glutamine. Remarkably, it has been reported that Ras-transformed cell lines utilize the macropinocytosis process to uptake extracellular protein into the cell. This mechanism of internalization and proteolytic degradation of protein restores the intracellular amino acid pool including glutamine. The authors suggested that it appears to support Ras-transformed cell metabolism when glutamine is scarce [41].
Related to tumor cell survival, recent observations have indicated that a population of dormant tumor cells remained, which are resistant after K-ras oncogene ablation (hereafter cited as surviving cells (SCs)) rely on OXPHOS activity for survival. After oncogene ablation, SCs rely less on glucose and glutamine and more on pyruvate and palmitate to restore TCA cycle intermediates [60]. However, the genetic or pharmacological inhibition of glutamine metabolism sensitizes tumors to ß-lapachone. This compound is an NADPH:quinone oxidoreductase that leads to NADPH and NAD+ depletion through the enhancement of ROS levels and hyperactivation of poly(ADP ribose) polymerase (PARP), respectively. As a consequence, combination therapy leads to cell death in oncogenic K-ras-expressing cells but not wild-type K-ras-expressing cells [61]. Together, these finding suggest that K-ras-mutant cell line-mediated activation of glutamine catabolism can provide rational targets to be novel anticancer therapies.
p53
Tumor suppressor genes have also been correlated with metabolic reprogramming in cancer. The most important tumor suppressor gene, known as p53 gene, encodes the transcription factor (p53). Expression is related to a wide array of stress stimuli through a complex cell signaling network. The role that p53 exerts on metabolic rewiring has been extensively investigated, but how p53 directly controls tumor cell metabolism remains to be elucidated [62]. Early studies found that p53-deficient cells increase glycolysis and decrease OXPHOS through the direct control of p53-inducible regulator of glycolysis and apoptosis (TIGAR) and cytochrome c oxidase (SCO2), respectively [63, 64]. Most recently, other observations have characterized a direct role of p53 by regulating the enzymes involved in glutamine metabolism such as GLS2 and malic enzymes (ME1 and ME2). Accordingly, activation of p53 seems to support tumor growth suppression and maintenance of senescence [65, 66]. The activation of p53 augmented GLS2 expression, resulting in enhanced levels of glutamate and α-KG, increased mitochondrial respiration/ATP production, and decreased ROS levels [65]. Moreover, p53 increased GLS2 expression in response to oxidative stress and accumulation of genome damage, reducing tumor formation [67]. p53 activation represses ME1 and ME2, decreasing glutamine metabolism, rather than glucose metabolism, leading to senescence [66]. Taken together, these findings demonstrate that the activation of p53 drives glutamine metabolism to restore cell homeostasis and tumor suppression through increased levels of GSH and decreased levels of ROS. Consistent with these observations, GLS2 expression was almost absent or significantly decreased in glioblastomas and overexpression of GLS2 was sufficient to strongly inhibit tumor progression [68]. Likewise, the correlation between p53 (that regulates GLS2), but not GLS1, and Myc that regulates GLS1 (but not GLS2) is tempting data to support the hypothesis that glutamine metabolism may be exploited for the discovery of therapeutic targets in different tumor cell lines which are genetically reprogrammed.
Surprisingly, recent reports have now highlighted a p53 signaling pathway that senses glutamine levels and mediates cell survival. The authors have found a serine/threonine protein phosphatase 2A-associated protein (PP2A) containing a regulatory subunit known as B55α that activates p53 through dephosphorylation of the domain-containing E3 ubiquitin ligase (EDD). Accordingly, the B55α-EDD-p53 pathway activation is mediated by ROS accumulation as a consequence of glutamine deprivation and is required for tumor growth [69]. Thus, although p53-mutant confers a selective growth advantage to cancer cells, loss of p53 impairs the ability of cancer cells to respond to nutrient deprivation. These features can be exploited to form therapeutic benefits to treat different human cancer cell lines, which harbor genetic alterations of TP53-encoding gene [70].
Sirt4 and Hace1
Others tumor suppressor genes have been reported to regulate glutamine metabolism, including Sirt4 and Hace1. Sirtuin is a family of NAD+-dependent deacetylases, deacylases, and ADP-ribosyltransferases proteins. Sirt4 is a mitochondrial ADP-ribosyltransferase protein involved in cell metabolism and aging [71]. Early evidence has connected the role of Sirt4 on glutamine metabolism as a potent inhibitor of GDH in pancreatic β cells [71]. More recently, GDH has been reported to be linked to DNA damage, glutamine metabolism, and SIRT4 in cancer [72]. After exposure of various cell lines to UV irradiation, the authors performed a time-course tracer study of [U-13C5]glutamine. They found a mechanism of DNA damage that drives Sirt4-mediated inhibition of glutamine anaplerosis necessary for cell cycle control. In addition, Sirt4-deficient fibroblasts have the ability to grow faster than their normal counterparts and developed lung tumors. Consistently, Sirt4 is a tumor suppressor acting as a repressor of the enzyme GDH, thereby blocking glutamine catabolism and cancer progression [73]. Remarkably, documenting a relationship between mTORC1 and Sirt4 has provided insights into tumor biology. Csibi et al. have showed that mTOR1 regulates GDH heightening glutamine catabolism (anaplerosis) through destabilization of the cAMP response element binding-2 (CREB2), which decreases Sirt4 expression [74].
Another tumor suppressor gene, known as Hace1 E3 ligase, is inactivated in a diverse range of human cancer cell lines. Hace1 E3 ligase was recently been identified to play an important role in glutamine metabolism. Hace1 is involved in decreasing the levels of superoxide and ROS level in vitro and in vivo. The interplay between glutamine and antioxidant mechanisms, such as NADPH and GSH, made it reasonable to test whether Hace1 mutantion alters glutamine metabolism. The authors showed that Hace1-mutant mouse embryonic fibroblasts (MEFs) were more sensitivity to glutamine withdrawal and exogenous inhibitors of glutamine metabolism as compared to normal counterparts. Hace1-mutant MEFs increased cellular sensitivity to ROS-associated apoptosis. Interestingly, treatment with the antioxidant N-acetylcysteine (NAC) or the TCA cycle intermediate OAA rescued cell survival. Accordingly, Hace1-mutant MEFs relied on glutamine uptake and NH4+ secretion to cope with the stress responses, including ROS production and microenvironment acidification [75].
Energy addiction, growth, and proliferation
The commitment of proliferating cells to drive anabolic needs (to support production of macromolecules) is consistent with the steady demand for increased energy addiction. To fine tune the high energy demands, tumor cells increase nutrients uptake like glucose and glutamine.
The most important energy signaling pathway is the PI3K/Akt/mTOR and is a common lesion in human cancer [76]. It is evident from many reports that the PI3K/Akt/mTOR pathway regulates glucose metabolism. Improved mitochondrial activity through the downstream control of mTORC1 [77–80] requires amino acids to promote protein synthesis, cell growth, and proliferation [8, 81]. Early evidence has suggested that the uptake of exogenous L-glutamine and its efflux out cells serves to maintain intracellular levels of essential amino acids (EAA) such as leucine, leading to the activation of the mTOR-S6K1 pathway. These findings provide further evidence that L-glutamine directly impacts on upstream mTORC1, leading to EAA exchange to regulate cell growth and proliferation [29]. Interestingly, intracellular levels of glutamine and leucine support glutaminolysis, promoting lysosomal translocation and mTOR activation [82]. Likewise, another report has demonstrated that mTORC1 activation regulates positively, GDH; this launches a robustly anabolic response through glutamine catabolism. Mechanistically, it has been shown to involve Sirt4/CREB2 repression/degradation (see “Sirt4 and Hace1”) [74]. Another study has demonstrated a key insight into the role of mTORC1 and Myc as central regulators of glutaminolysis. The authors have shown that mTORC1 regulates GLS1 expression, but not GLS2, through S6K1/eIF4B substrates via Myc-dependent activation. They also suggested the rationale of targeting glutamine metabolism as a cancer therapy [9]. Thus, glutamine catabolism is directly controlled by mTOR activation.
In addition, recent work has demonstrated that glutamine seems to act coordinately to control mTOR activation and inflammation. Accordingly, when glutamine was scarce in the cell culture medium, it induced an endoplasmic reticulum (ER) stress-response consistent with the induction of expression and secretion of chemokines (e.g., interleukin-8 (IL-8)). As a result, short-term glutamine restriction activated the mTOR-IRE1-JNK pathway and strongly enhanced the autophagy response. Interestingly, IL-8 secretion was autophagy-independent and it has successfully correlated with deficient glutamine-anaplerosis precursors, since dimethyl alpha-KG (DM-α-KG) supplementation was sufficient to abrogate IL-8 secretion. The authors have also suggested that the decrease of TCA intermediaries after glutamine deprivation may be consistent with increased ROS levels, whereas the treatment with NAC reduced IL-8 secretion [83]. These findings are consistent with our observations, in which tumor cell lines when subjected to high exposure of nitric oxide (NO) or hydrogen peroxide (H2O2) activated their transcriptional profile, favoring pro-inflammatory signaling. As a result, these cell lines became radioresistance, as well as resistance to many anticancer agents when compared with their corresponding parent cell lines [84, 85]. Shanware et al. further developed the idea that potential inhibitors of glutamine metabolism, including compound 968, bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES), and glutamine analogous (see “Targeting glutamine metabolism” for therapy) might also be combined with antagonists of chemokines for therapeutic purposes to treat radioresistance cancer cells [83].
Current models describe how non amino acid availability inhibits mTOR activity and how autophagy is directly activated. In this regard, autophagy is activated to recycle macromolecules through the lysosome for subsequent fuel bioenergetic programs [86]. Remarkably, Palm et al. showed that the mTORC1 pathway has opposing effects on cell proliferation depending on the amino acid source available. For example, during nutrient-depleted conditions, the inhibition of mTORC1 results in increased catabolism of endocytosed proteins and autophagy, enhancing cell proliferation [87]. Therefore, taken in consideration of the contribution of glutaminolysis to autophagy, which was not consistent with nutrient deprivation and mTOR inhibition, it was dependent on ammonia production. These data may provide key insights in the current mechanism of interplay between glutamine and cell signaling in cancer that needs to be further investigated [31, 83]. We address this specific topic below (see “Depot for ammonia and autophagy”).
Another important pathway extensively studied in cancer is the AMP-activated Ser/Thr protein kinase pathway (AMPK). It is evident from many reports that AMPK serves as a cellular energy sensor, which regulates metabolism to maintain cellular energy homeostasis. In low-energy states, AMPK coordinates many metabolic pathways by favoring catabolism to produce energy while it shuts down key anabolic bioenergetics programs such as the synthesis of simple molecules and macromolecules [88]. To efficiently preserve energy, AMPK is capable to activate autophagy, to inhibit lipogenesis, and to block mTORC1 activation and subsequent protein biosynthesis [89–91]. Emerging observations argue for a more profound metabolic function of AMPK. AMPK activation is sufficient to repress aerobic glycolysis in tumor cell lines and inhibits tumor growth in vivo. Clearly, the authors showed the reallocation of glucose into the biomass (e.g., amino acids and lipids) when AMPK was inactivated, suggesting that AMPK plays a role in preventing cancer progression [92]. However, AMPK activation under nutrient deprivation or in tumor cell lines genetically programmed by oncogenes can maintain metabolic homeostasis and survive [93, 94]. It explains in part the interplay between glutamine metabolism and AMPK, despite the fact that the full mechanism is not currently understood. Observations like these have showed that human and murine cell lines with Myc-derangement expression are dependent on AMPK-related kinase 5 (ARK5), enabling AMPK activation, and mTORC1 inhibition. The ARK5 signaling was required to enhance energy production via glutaminolysis, providing intermediates for the TCA cycle. As a result, tumor cells are able to maintain mitochondrial integrity and respiratory capacity [93].
Another important pathway involved in adaptive response to nutrient stress is the protein kinase C. It has been reported that an unexpected metabolic reprogramming of protein kinase C (PKCζ)-deficient human cancer cells that increase the glutamine metabolism, during glucose starvation, through the serine biosynthetic pathway, leading to a rise in the antioxidant defense. PKCζ represses the expression of either PHGDH and PSAT1 enzymes, diminishing or abolishing the flux of 3-phospoglycerate to serine formation, and it could be the reason of why PKCζ acts as a tumor suppressor. These findings suggest that malignant cells exhibit metabolic flexibility to sustain growth and survival [94].
Although the role of glutamine in cancer progression has been extensively studied over recent years, more findings may be meaningful to understand the interplay between glutamine and the cell signaling response (e.g., mTORC1, AMPK, and other protein kinases) in a wide range of stimuli and in a specific cellular context.
Oncometabolites for synthesis of biomass and malignancy
As previously detailed, tumor cells rely on glucose and glutamine metabolism to grow. It is likely that glutamine metabolism along with aerobic glycolysis can also support building block synthesis (e.g., amino acids, lipids, and nucleotides), redox balance, energy production, and cell signaling in cancer. In tumor cells, the glycolytic flux is tightly coordinated by HIF1-α, which regulates the expression of a variety of enzymes involved in glucose metabolism, including Glut1, HKs, PFK2, aldolase, PGM, PKM2, LDHA, PDK1, and MCT4 [44]. The excess consumption of glucose is due in part to generate ATP via glycolysis, although this process is an important source of biosynthetic precursors rather than a source of ATP [95]. Intriguingly, to optimize bioenergetics demand, hematopoietic cells coupled glutamine consumption to glucose availability, while K-ras transformed NIH3T3 fibroblast decoupled glucose and glutamine metabolism to sustain cell growth [32, 58]. Here, we highlight the role of the most important glycolytic and glutaminolytic enzymes involved in cancer progression and how these enzymes might coordinate cell metabolism during the transformation process.
It is reasonable to think that the “TCA cycle clockwise” happens when cancer cells need a high demand of ATP, to coordinate a preparation phase for proliferation, as well as the production of GSH, since the DNA will be exposed to ROS and RNS, which are linked with DNA mutations in cancer [96–98]. Tu et al. showed an interesting pattern of metabolite accumulation during periodic phases of Saccharomyces cerevisiae [99]. They observed that numerous amino acid precursors and amino acids, nucleotide precursors (e.g., IMP), and TCA cycle intermediates (e.g., isocitrate, succinate, and pyruvate) increased significantly during the oxidative phase, confirming the anaplerotic role of the TCA cycle clockwise, in which NADH production is exacerbated to be used into the electron transport system. On the other hand, during the reductive/building phase, a high abundance of metabolites like pyruvate and glucose-6-phosphate indicated the transition from oxidative to glycolitic metabolism, reducing the risk of DNA damage through oxidative stress. Notably, it happens if cancer cells have dynamic TCA cycle operating and a functional complex I [100]. However, many tumor cells have been discovered with mutations in genes that encode TCA cycle metabolic enzymes, including succinate dehydrogenase (SDHB, SDHC, and SDHD), fumarate hydratase (FH), and mitochondrial isocitrate dehydrogenase (IDH1 and IDH2) [40]. How can mutations in metabolic enzymes favor the cell transformation process? Techniques have improved in isotopic labeling of metabolites. Thus, it is becoming possible to analyze in detail the metabolic rewiring of tumor cells. For instance, both SDH and FH are key enzymes of the TCA cycle that convert succinate to fumarate and fumarate to malate, respectively. Early studies have described mutations in subunits B, C, and D of SDH and mutations in FH, which are associated with the development of paraganglioma and renal cell carcinoma [101]. These mutations contribute with the accumulation of oncometabolites (such as succinate and fumarate which are released into the cytosol), where they contribute to the inhibition of the prolyl hydroxylase enzymes (PHDs) [102]. Of note, the inhibition of PHDs leads to stabilization of HIF1α, causing a tumorigenic pseudo-hypoxia state. Recently, it was proposed that the accumulations of both succinate and fumarate drive oncogenesis through the inhibition of α-KG-dependent histone and DNA demethylases, leading to subsequent alterations of genome-wide histone and DNA methylation [103]. Intriguingly, it has been demonstrated that modified mouse kidney Fh1-deficient cells increase in the glutamine uptake. These Fh1-deficient cells use significantly more glutamine than Fh-normal cells through the accumulated TCA cycle metabolites, leading to partial mitochondrial NADH production [104].
The massive elevation of 2-HG in tumors with mutated IDH1 (cytosolic) or IDH2 (mitochondrial) has shown a reductive pathway of glutamine metabolism dependence to survival. Moreover, genetically suppressed expression of the pyruvate dehydrogenase A1 (PDHA1) relies on the reductive carboxylation branch of glutamine metabolism through IDH1 to generate lipogenic acetyl-CoA [105]. In this context, glutamine is the major lipogenic precursor for cancer cell growth. It plays a key role to restore the intracellular redox homeostasis, rendering an oxidative function [37]. Moreover, the accumulation of 2-HG has been linked to modulate gene expression in cancer cells through epigenetic modifications, including inhibition of DNA and histone demethylases, and activation of prolyl hydroxylases, which are consistent with cell transformation [39, 106–108].
This noncanonical mechanism facilitates the reductive carboxylation from α-KG to isocitrate by mediation of IDH2 and IDH3, and then, by the activity of aconitase, it can be transformed to citrate. It is interesting to note that IDH2 uses NADPH as cofactor and IDH3 uses NADH. Thus, reductive carboxylation can act as a regulator of the redox balance, by maintaining the NADH/NADPH ratio. When citrate accumulates, it is exported from mitochondria to the cytoplasm, where it could be converted into OAA and AcCoA by the activity of ATP citrate lyase (ACLY). ACLY activity relies on ATP as a rate-limiting cofactor; therefore, when cancer cells have a high concentration of ATP and NADPH, this enzyme favor lipid biosynthesis and cell proliferation [109]. Interestingly, the inhibition of ACLY can decrease tumor growth [110]. Furthermore, OAA can be transformed to aspartate for protein synthesis, or can be fueled to generate lactate. This set of reactions are consistent with the conversion of OAA to malate by the cytosplasmatic malate dehydrogenase, then to pyruvate by ME1, and finally to lactate by LDHA. Lactate is considered another important oncometabolite that plays a key role in cancer. It is known that lactate can be produced from glucose metabolism, glutamine metabolism, and serine metabolism, but the understanding of the intermediary metabolism repertoire used for tumor cells to produce the same metabolite remains under strong investigation in cancer biology. The vast literature on cancer biology has pointed out the role of lactate as an oncometabolite generated by “the Warburg effect.” More recently, using advanced labeling techniques, it has been demonstrated in glioblastoma cells that the lactate produced by glutaminolysis was necessary to increase NADPH, supporting fatty acid synthesis [17]. Intriguingly, cancer cells responded to serine deprivation by decreasing lactate production, where serine takes place as an allosteric activator of PKM2 [111]. For example, glucose limitation elicits an increased metabolic flux from glutamine metabolism into the serine and glycine biosynthetic pathway [112]. In this context, some studies have addressed the key role of some metabolic enzymes that participated directly or not, on the gluconeogenesis; for example, phosphoenol pyruvate carboxylase kinase (PEPCK) activation or PFKFB3 inhibition. These enzymes are important steps required to control metabolism of tumor cells during nutrient deprivation [113, 114]. Overall, these data suggest that tumor cells have much more complex metabolic repertoire than previous appreciated, and that some tumor cells are reliant on glutamine toward anabolic metabolism. As we have mentioned, the glutamine catabolism drives the ATP production by the TCA cycle clockwise, and proliferation by reductive carboxylation. An important by-product of this process is citrate, which has a pivotal role in glycolysis. When ATP accumulates, it inhibits a phosphofructokinase (PFK), a rate-limiting enzyme of the glycolytic pathway. PFK inhibition is enhanced by citrate, which diminishes the glycolytic flux [115], and sustains the uptake and utilization of glutamine.
Many studies have focused on nutrient deprivation to understand the metabolic flexibility displayed by cancer cells. For example, when glucose is scarce, cells adapt by switching to glutamine as a carbon source, but when cells growing during glutamine shortage, glucose becomes the major nutrient source. As previously mentioned, it has been proposed that phosphoenolpyruvate carboxykinase (PEPCK), the rate-limiting enzyme of gluconeogenesis (that catalyzes the conversion of OAA into phosphoenolpyruvate) increases glucose and glutamine utilization toward anabolic metabolism, leading to cancer cell proliferation. There are two isoforms, PEPCK1 or PCK1 (cytosolic) and PEPCK2 or PCK2 (mitochondrial). Interestingly, it has been shown that PCK2 regulates gluconeogenic process in nonsmall cell lung cancer (NSCLC), using carbons derived from glutamine to produce biosynthetic intermediates. They demonstrated that glutaminolysis can support citrate production and serine biosynthetic pathway during glucose restriction. Montal et al. studying the PCK expression in colon cancer samples, found PCK1 increased, but not PCK2. After silencing PCK1, they found a decrease of intermediary TCA’s metabolites and the regression of the tumor volume. Unexpectedly, they found the carbons derived from glutamine in pentose phosphate pathway (PPP), and the production of ribose from glutamine, elucidating a new anabolic pathway [113]. Taken together, PEPCK is a central enzyme that connects carbon metabolism. Coordinating metabolism of glucose, glutamine, and other amino acids, as well as PEPCK directly regulates the TCA cycle flux and cell proliferation. As such, PEPCK’s role can be exploited for cancer therapy. Most recently, the role of serine and glycine has been appreciated in cancer [116]. Cancer cells uptake glucose and glutamine to produce serine and glycine, using intermediate metabolites of glycolysis and glutaminolysis. For instance, 3PG, a glycolytic metabolite, is converted to 3-phosphopyruvate by PHGDH, and it is transaminated with glutamate (mainly produced by the glutaminolytic pathway) to produce 3-phosphoserine, and consequently, serine and glycine. The PHGDH expression is frequently upregulated in melanoma and triple negative breast cancers [117]. The serine biosynthetic pathway is a crucial point in glucose metabolism [118]. Cancer cells can consume glycine through the one carbon metabolism, producing nucleotides, lipids, proteins, and other cofactors, or using it as an anaplerotic fuel of the TCA cycle and antioxidant defense. These nutrients can be used for the biosysnthesis of proteins, contributing to cancer cell metabolic autonomy [117, 119]. In 2013, Maddocks et al. showed that p53 is related to serine starvation and oxidative stress, preserving the cellular antioxidant capacity. Cancer cells lacking p53 reduced their viability and proliferation [120]. Recently, the p53-family member p73 has been linked to play an important role in the serine biosynthesis, under metabolic stress [121]. The glycine generated by the serine metabolism is related to maintain DNA stability and normal methylation patterns [122, 123]. Related effects of folate metabolism were reported by Farber and Diamond in 1948, after treatment of children with AML by the treatment with 4-aminopteroylglutamic acid, leading to a temporary remission of leukemia [124]. In this regard, it is important to understand why serine metabolism is important to connect glucose and glutamine metabolism with survival, growth, and energetic processes.
Cellular redox balance and stress resistance
The etiology of breast, prostate, pancreatic, and colon cancer has been correlated with oxidative stress, mainly by the interactions with the complex tumor environment [125]. Oxidative stress is a result from the imbalance between ROS levels and antioxidant mechanisms. In cell physiology, there are various endogenous sources of ROS, including mitochondria, the NADPH oxidase, the cytochrome P450 system, the xanthine oxidase, and the nitric oxide synthase [126]. The presence of diverse antioxidant defenses, the so-called phase II detoxification enzymes, such as the glutathione reductase (GR), the glutathione-S-transferase (GST), the glutathione peroxidases (GPx), and many others, are there to counteract the ROS produced both in and outside the cells. The loss-of-function of antioxidant enzymes coupled with the excess of ROS are sufficient to promote DNA, lipid, protein, and membrane damage leading to tumor transformation [127–129]. Here, we address the main role of glutamine in redox homeostasis.
It is interesting to delineate that “the Warburg phenotype” offers an alternative defense mechanism, since it can reprogram the metabolism to citrate formation used for biomass synthesis, resulting in less metabolites to replenish the TCA cycle and, consequently, OXPHOS [4]. However, cancer cells that incorporate higher amounts of glucose and glutamine rely on GSH biosynthesis, the major antioxidant defense, to resist against oxidative stress. As illustrated in Fig. 1, GSH is produced when the cytosol is fueled by glutamate, cysteine, and glycine. Glutamine is converted to glutamate, and the glutamate pool is necessary to acquire cysteine through a mechanism which exports glutamate and imports cystine. In this regard, glutamine increases GSH metabolism for maintaining the cellular GSH/GSSG ratio by a wide range of stress stimuli [130, 131]. It has been demonstrated that malignant tumor cells maintain a supply of GSH when compared with their normal counterparts to resist the oxidative stress generated through metabolic abnormalities [132].
Another example, nuclear factor-erythroid 2 related factor 2 (Nrf2) has been related to support cancer chemoresistance [133–135]. Nrf2 is a redox-sensitive factor that binds to antioxidant response element (ARE), regulating the expression of SOD, catalase, and HO-1 [136]. Overexpression and hyperactivation of Nrf2 may play an important role in tumorigenesis, enabling Fh-mutant cells to tolerate high levels of oxidants [104]. One explanation is that Nrf2 activates the expression of genes involved in glucose metabolism and glutamine metabolism, including intermediates of the PPP and glutaminolysis [137]. Correlated with the ability of tumor cells to coordinate the most important nutrient pathways to support proliferation, inhibiting antioxidant capacity of these cells may be a useful therapeutic approach. Recently, DeNicola et al. showed that Nrf2 regulates serine and glycine biosynthetic pathway in NSCLC. Using 13C labeled glucose, they found an accumulation of serine M3 13C labeled and glycine M2 13C in cells with high expression of Nrf2. Genetic inhibition of Nrf2 decreased the mRNA expression of the serine biosynthesis pathway’s enzymes, showing the role of Nrf2 as a regulator of this pathway, via activating transcription factor 4 (ATF4), a direct transcriptional target of Nrf2 [138] and/or heterodimerization partner of Nrf2 [139]. This pathway regulates glutathione formation through the cysteine and glycine labeled from glucose [140], demonstrating the role of Nrf2 on the interplay between glutamine consumption and glucose utilization through the serine biosynthetic pathway.
With the increasing efforts to understand the glutamine metabolism in cancer cell culture, there is an increasing need to address the role of the redox homeostasis. Aberrant oncogenic or tumor suppressor signaling (see “Oncogenic and tumor suppressor signaling pathways” section), hypoxia, metabolic defects, and ER-stress induce ROS and increases the demand for robust systems to establish the cellular redox status. Conversely, levels of ROS are eliminated by the production of glutathione and NADPH as well as ROS scavengers through Nrf2 [141, 142]. As well described here, glutamine metabolism has been shown to be crucial to regulate cellular redox status, contributing to tumor development. A number of studies have shown that glutaminase enzymes play an important role in regulating intracellular levels of ROS. For instance, genetic downregulation of either GLS2 or p53 compromises the GSH-dependent antioxidant system and increases intracellular ROS levels [67]; however, their activation increase GSH levels and decreases ROS levels, leading to cancer survival [65]. On the other hand, glutamine-fueled mitochondrial metabolism generates ROS by OXPHOS activity, leading to cell proliferation and tumorigenesis [57, 143]. These conflicting effects of ROS have important implications for novel anticancer therapy. In fact, the dual function of ROS including both the physiological and pathological remains as a challenge to target cancer therapy. Thus, more studies would be needed to define whether to use pro-oxidant therapy to favor physiological ROS responses or antioxidant therapy to avoid ROS pathologies, which would help to solve this central question in redox biology [142].
Depot for ammonia and autophagy
Glutamine per se plays a pivotal role in supporting anaplerosis and redox balance through an anabolic response that supports energy production and cell biomass in proliferating cells [17, 144]. Not surprisingly, however, proliferating cells that rely on glutaminolysis might become a depot for NH4+. The role of ammoniagenesis in cancer has recently been investigated, although the exact mechanism remains unknown. Traditionally, ammonia was considered to be a potentially cytotoxic by-product. Most recently, Yang et al. have demonstrated that GLS and GDH generate NH4+ from both γ- and α-nitrogens of glutamine, respectively. Studying glioblastoma cells with enhanced Myc activity through GC/MS techniques, the author reported that after 8 h, more than 90 % of the ammonia secreted in the medium was derived from both nitrogens [36]. But, later studies on tumor cells revealed that glutamine consumption/ammonia secretion ratio is about 75 % [33].
Early observations showed that ammonia has been linked to the regulation of metabolic processes, such as long-term survival and proliferation of hepatic 3B when glutamine is depleted [145]. However, the idea that ammonia generated during glutaminolysis could presumably modulate macromolecule turnover by autophagy activity boosted the interest in studying ammoniagenesis and cancer metabolic rewiring. The authors have previously showed that after 2 days of cell culture, there is substantially enhanced autophagy activity. Surprisingly, this process was nutrient deprivation-independent and independent of mTOR inhibition. As a result, the content medium from these cells was analyzed and the authors indentified ammonia as a direct factor that triggered autophagy in secondary cell cultures. Furthermore, ammonia protected these cells from tumor necrosis factor α (TNF-α) [31]. One explanation for this remarkable result is that in a stressful tumor microenvironment, when glutamine is an essential nutrient for tumor growth, ammonia triggers autophagy activity as a master signal. This allows tumor cells increase survival by effectively recycling macromolecules [146]. Nonetheless, as ammonia is a diffusible by-product of glutaminolysis, it can trigger autophagy in tumor areas not well vascularized [147]. Interestingly, recent work demonstrated that ammonia induce autophagy using the common autophagic program (e.g., Atg8 and Atg5), but was independently of ULK1/ULK2 kinases; a downstream signal of the mTOR pathway. Studies of MEFs deficient for ULK1 and ULK2 have shown that this canonical autophagy-axis was not required to activate autophagy during prolonged glucose starvation or when cells were challenged with ammonia. This suggesting that ammonia must be considered as a key metabolite to adapt cells to meet there nitrogen demands [47]. Together, these finding showed an unknown ammonia-dependent pathway involved in autophagy. Although the exact molecular mechanism is not fully understood, if ammonia-induced autophagy has a cellular-protective role in cancer, then the inhibition of autophagy may be a therapeutic approach to treat cancer patients [148].
Glutamine metabolism as a target for cancer therapy
Targeting glutamine metabolism
Several studies, using tumor cell lines, have demonstrated that mitochondria operate as a major player in regulating cellular bioenergetics to respond to the metabolic demands of the cell. In this regard, mitochondria can determine the cell fate between life and death via apoptosis. Thus, it is becoming more feasible that mitochondria can be exploited for cancer therapeutic approaches. However, there are still some misconceptions to be elucidated, such as the relation between cell death regulation and mitochondrial metabolism [149]. Currently, three different manners have been described to target mitochondria: (i) targeting mitochondrial bioenergetics, (ii) mitochondrial biosynthetic production, and (iii) mitochondrial redox balance [150]. As mentioned above, mitochondrial glutamine metabolism could exert its effect to benefit cell biomass (e.g., nucleotides, proteins, and lipids), redox balance (e.g., NADPH), and bioenergetics to energy production. Herein, we will now focus on glutamine metabolism in order to review how there can be so many ways to exploit the mitochondrial biosynthetic production and its promise to target cancer.
Certain cancer cell lines rely on glutamine metabolism for growth and survival, including pancreatic cancer, glioblastoma multiforme, leukemia, lung cancer, and breast cancer. Glutamine analogues, such as 6-diazo-5-oxo-L-norleucine (L-DON) and azaserine, have been found to decrease tumor cell proliferation, both in vitro and in vivo [151]. Subsequent experiments using the SLC1A5 transporter inhibitor γ-L-glutamylp-nitroanilide (GPNA) have suggested that the suppression of glutamine uptake also reduces tumor growth and mTOR activation [26, 29]. As previously mentioned, glutaminolysis plays an important role in cancer. Numerous reports have recently explored both rate-limiting glutaminolysis-enzymes: GLS1 and GDH. For instance, using the GLS1 enzyme inhibitor, compound 968, it was demonstrated to be a strong inhibitor of tumor growth [46]. Other reports using another specific GLS1 inhibitor (BPTES) showed great efficacy to slow growth of glioblastoma-associated IDH1 mutant, as well as inhibited the proliferation of many glutamine-addicted cancer cell types [13, 152, 153]. In addition, other reports have revealed the effect of innumerous glutamine metabolism inhibitors. For example, aminooxyacetate (OAA), the nonspecific inhibitor of aminotransferase (TA) enzymes showed a cytostatic effect on proliferating breast cancer cell lines [154]. Furthermore, epigallocatechin gallate (EGCG), the inhibitor of GDH, demonstrated great efficacy to kill glutamine-addicted tumor cells when glucose was scarce [36]. In general, inhibitors that block glutaminolysis were sufficient to delayed tumor growth in a number of models, both in vitro and in vivo [36, 48]. Most recently, a class of allosteric inhibitors of glutaminase C (isoform GAC) has been documented to interfere specifically with the GLS isoform GAC activity, providing insight into the glutamine metabolism and tumor progression [155].
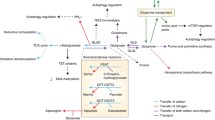
Another promising approach, is the possibility to disrupt glutamine metabolism with L-asparaginase; a wide spread pharmacologic compound used to treat pediatric acute lymphoblastic leukemia (ALL) [156]. The effect of L-asparaginase has been associated with glutamine depletion, and it appears to be an efficient strategy to exploit glutamine-addicted tumor cells. It is important to note that targeting glutamine metabolism in cancer may lead to new therapeutic approaches (see Fig. 2). The challenge is to exploit glutamine metabolism and combine it with other potential drugs already registered with the Food and Drug Administration (FDA), including mitochondrial complex I inhibitors (e.g., metformin and phenformin), modulators of autophagy (e.g., chloroquine and hydroxychloroquine), and inflammatory agents (e.g., verinostat) [157]. In fact, the increased glutamine “addiction” of some cancers has been exploited in the clinic as a way to disrupt the tumor growth. For example, CB-839 that is a derivative compound of BPTES has been developed and is currently being administered to humans in phase I clinical trials for solid tumors, lymphoid, and myeloid malignancies (NCT02071862, NCT02071888, and NCT02071927, respectively) [158]. Another compound known as fenretinide is currently in clinical trials to treat neuroblastoma (NCT00646230), B cell lymphoma (NCT00288067), and ovarian cancer (NCT01535157). This compound has been shown efficacy in Myc-amplified lymphoma and neuroblastoma during glutamine limitation, triggering apoptosis via the activation of the ATF4-PUMA/NOXA/TRB3 axis [159]. To note, the inhibitor of GDH enzyme (EGCG) has been used in combination with other drugs to treat squamous cell carcinoma of the head and neck (SCCHN) and has shown remarkable potential as chemopreventive agent [160]. Phenylbutyrate (AZ 85258-2463) is another agent that has been used in combination with fluorouracil in phase I–II studies of patients with advanced ovarian cancer. Phenylbutyrate depletes plasma glutamine and has shown low toxicity but also low efficacy. Clinical trials involving CB-839, phenylbutyrate, and EGCG-mediated glutamine metabolism inhibition for cancer therapy are ongoing and listed at http://www.clinicaltrial.gov.

The potential targets for cancer therapy. Many types of cancer rely on glutamine and glucose to support survival and growth. Myc and K-ras activate the expression of genes involved in glutamine metabolism, including SLC1A5, GLS1, and GDH. SIRT4 is a potent inhibitor of glutaminolysis by repressing the expression of GDH. Evidence also suggests a role for targeting glutamine metabolism by (a) using a glutamine competitor (e.g., L-asparaginase), (b) glutamine analogues (e.g., L-DON), (c) exogenous inhibitors of glutaminolysis (e.g., C968, BPTES, and EGCG), (d) specific inhibitor of glutamine transporter (e.g., SLC1A5), (e) nonspecific inhibitor of transaminases (e.g., AOA), or (f) oncometabolite analogs (e.g., 2HG-DH). Targeting glutamine metabolism has demonstrated therapeutic efficacy in a number of models of glutamine-addicted cancer cell types. Innumerous reports have used 18F-labeled analogs of glutamine that serves as a metabolic marker for the localization of tumor tissue (see below)
Measuring glutamine in cancer
As mentioned above, the clinical procedure of glucose-based imaging by 18F-FDG PET has been extensively applied for cancer diagnostic purposes [4]. Since emerging data has pointed out the dependence of certain tumor cell types to glutamine consumption, there is renewed attention to measure glutamine in cancer. In this regard, innumerous studies have identified some analogs of glutamine as potential tracer to identify and characterize tumor cell types that rely on glutamine metabolism. For example, the new tracer L-[5-11C]-glutamine was developed to serve as a metabolic marker for probing glutamine-addicted tumors not detected by 18F-FDG PET [161]. Studies with glioblastoma cells with enhanced Myc activity identified another analog of glutamine (8F-(2S,4R)4-fluoroglutamine). Subsequent in vitro and in vivo experiments have suggested that the monitoring of glutaminolysis could be exploited to generate imaging tracer diagnostics to benefit cancer patients [162, 163]. Currently, there have been a number of clinical trials in patients using different radioisotopes, like 11C, 13N, or 18F to image glutamine [16].
Moreover, detailed metabolic maps have been extensively studied in many cancer cell types [164]. Probing metabolic pathways using stable isotopes to trace single labeled [1-13C]glutamine or [5-13C]glutamine combined with uniformly labeled [U-13C5] glutamine, have allowed precise nutrient uptake and metabolite secretion of glutamine. From these studies, it was found that glutamine can contribute into numerous pathways [17]. For instance, labeling on all five carbons of glutamine is sufficient to recognize how cells allocate glutamine into the oxidative or reductive TCA metabolism. Furthermore, when glutamine is labeled with one radioactive carbon, it can be used to measure and quantify single intermediates metabolites downstream glutamine catabolism, including aspartate, citrate, malate, fumarate, and others [37, 38, 165]. In addition, uniformly labeled [U-13C5]glutamine has been applied to study tumor metabolism in mouse xenograft models [166]. Stable isotope analysis has been applied in a wide spread manner to quantify metabolites within cells, tissues, and plasma samples [164].
Conclusion, remaining questions, and perspectives
This review has highlighted the emerging insights regarding glutamine metabolism in cancer. Glutamine is one of the most abundant amino acid present in the plasma that serves to sustain bioenergetics, cell biomass, redox balance, and nitrogen balance. Moreover, as mentioned above, several oncogenes and/or tumor suppressor genes have been described to rely directly on, or at least in part, on glutamine metabolism. Cell lines with increased Myc-expression rely on glutaminolysis and NH4+ release, while K-ras-driven cancer cells rely on glutamine through transamination to fuel their energy needs [167]. Targeting glutamine catabolism and anabolism for cancer therapy is emerging as a feasible idea to study and treat cancer. One substantial consideration is that not all cancer cell types depend on exogenous glutamine. In some circumstances, such as in glutamine-free media, it has also been shown that certain cancer cell types can also display a plasticity to take up different amino acids, including serine, glutamate, asparagine, proline, and aspartate or recycling extracellular protein to support their energy supply and macromolecules needs. There remain three major challenges to fully understand cancer metabolism: (i) urge a standardization of cell culture media with complete protocols defining media, such that mechanism as macropinocytosis can scavenge extracellular protein to support K-ras-driven cell proliferation in glutamine depletion condition [168]; (ii) point out how and which nutrients are intertwined in maintain cell survival, and in which specific cellular context, regarding ambiguous results that have been generated when studying heterogeneous cancer cell populations and cell compartmentalization; and (iii) link how mitochondrial bioenergetics and metabolism can be reprogrammable in various cellular contexts—for instance in immune cells, stem cells, neuronal cells, and proliferative cells. Furthermore, targeting mitochondria to induce cell death via apoptosis should be linked with other types of cell death programs, including mitotic catastrophe, autophagy, and necrosis. Understanding glutamine metabolism will allow the development of novel therapies based on the peculiar features of mitochondrial bioenergetics in cancer.
Abbreviations
- 2-HG:
-
2-Hydroxyglutarate
- 13CNMR:
-
Magnetic resonance spectroscopy
- α-KG:
-
α-Ketoglutarate
- AMPK:
-
AMP-activated Ser/Thr protein kinase
- AOA:
-
Aminooxyacetate
- BPTES:
-
Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide
- CS:
-
Citrate synthase
- EAA:
-
Essential amino acids
- ER:
-
Endoplasmic reticulum
- ETC:
-
Electron transfer chain
- FADH2 :
-
II flavin adenine dinucleotide
- FH:
-
Fumarate hydratase
- GDH:
-
Glutamate dehydrogenase
- GLS:
-
Glutaminase
- GOT1:
-
Aspartate transaminase
- GPNA:
-
γ-L-glutamylp-nitroanilide
- GS:
-
Glutamine synthetase (GS)
- GSH:
-
Glutathione
- GSSG:
-
Glutathione disulfide, Hace1, E3 ubiquitin-protein ligase
- HIF-1:
-
Hypoxia-inducible factor-1
- HBP:
-
Hexosamine biosynthetic pathway
- IDH:
-
Isocitrate dehydrogenase
- LDH:
-
Lactate dehydrogenase
- L-DON:
-
6-Diazo-5-oxo-l-norleucine
- ME:
-
Malic enzyme
- mTOR:
-
Mammalian target of rapamycin
- NADH:
-
Nicotinamide adenine dinucleotide
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NEAA:
-
Nonessential amino acids
- NH4+ :
-
Ammonia
- NO:
-
Nitric oxide
- OAA:
-
Oxalacetate
- OXPHOS:
-
Oxidative phosphorylation
- PARP:
-
Poly(ADP ribose) polymerase
- PC:
-
Pyruvate carboxylase
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PEPCK:
-
Phosphoenol pyruvate carboxylase kinase
- PDH:
-
Pyruvate dehydrogenase
- PHD:
-
Prolyl hydroxylase
- PHGDH:
-
Phosphoglycerate dehydrogenase
- PK:
-
Pyruvate kinase
- PSAT1:
-
Phosphoserine aminotransferase
- ROS:
-
Reactive oxygen species
- SCs:
-
Surviving cells
- SCO2:
-
Cytochrome c oxidase
- SDH:
-
Succinate dehydrogenase
- Sirt4:
-
Mitochondrial ADP-ribosyltransferase
- SLC1A5:
-
Solute linked carrier family 1 member A5
- TA:
-
Aminotransferase
- TCA:
-
Tricarboxylic acid
- TDG:
-
Thymine DNA glycosylase
- TNF-α:
-
Tumor necrosis factor α
References
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Warburg O, Posener K. Ueber den Stoffwechsel der Carcinomzelle. Naturwissenschaften. 1924;12:1131–7.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–9.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
DeBerardinis R, Sayed N, Ditsworth D, Thompson C. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18(1):54–61.
Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12(11):829–46.
Vander Heiden MG. Exploiting tumor metabolism: challenges for clinical translation. J Clin Invest. 2013;123(9):3648–51.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308.
Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24(19):2274–80.
Gaglio D, Soldati C, Vanoni M, Alberghina L, Chiaradonna F. Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS One. 2009;4(3), e4715.
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–5.
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481(7381):380–4.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–5.
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782–7.
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178(1):93–105.
Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123(9):3678–84.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345–50.
Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. 2015;17(12):1556–68.
Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol. 2014;10:728.
Zhang J, Fan J, Venneti S, Cross J, Takagi T, Bhinder B, et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell. 2014;56(2):205–18.
Eagle H. Nutrition needs of mammalian cells in tissue culture. Science. 1955;122(3168):501–14.
Sauer L, Stayman J, Dauchy R. Amino acid, glucose, and lactic acid utilization in vivo by rat tumors. Cancer Res. 1982;2(10):4090–7.
Bergström J, Fürst P, Norée L, Vinnars E. Intracellular free amino acid concentration in human muscle tissue. J Appl Physiol. 1974;36(6):693–7.
Reitzer L, Wice B, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254(8):2669–76.
Witte D, Ali N, Carlson N, Younes M. Overexpression of the neutral amino acid transporter ASCT2 in human colorectal adenocarcinoma. Anticancer Res. 2002;22(5):2555–7.
Hassanein M, Hoeksema M, Shiota M, Qian J, Harris B, Chen H, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013;9(3):560–70.
Huang F, Zhao Y, Zhao J, Wu S, Jiang Y, Ma H, et al. Upregulated SLC1A5 promotes cell growth and survival in colorectal cancer. Int J Clin Exp Pathol. 2014;7(9):6006–14.
Wang Q, Hardie R, Hoy A, van Geldermalsen M, Gao D, Fazli L, et al. Targeting ASCT2-Mediated Glutamine Uptake Blocks Prostate Cancer Growth and Tumour Development. J Pathol. 2015. doi:10.1002/path.4518.
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136(3):521–34.
Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell. 2015;9(27):354–69.
Eng CH, Yu K, Lucas J, White E, Abraham RT. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci Signal. 2010;3(119):ra31.
Wellen K, Lu C, Mancuso A, Lemons J, Ryczko M, Dennis J, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24(24):2784–99.
DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29(3):313–24.
Bröer A, Wagner C, Lang F, Bröer S. The heterodimeric amino acid transporter 4F2hc/y+LAT2 mediates arginine efflux in exchange with glutamine. Biochem J. 2000;1(349):787–95.
Timmerman L, Holton T, Yuneva M, Louie R, Padró M, Daemen A, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–65.
Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69(20):7986–93.
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–8.
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108(49):19611–6.
Dang L, White D, Gross S, Bennett B, Bittinger M, Driggers E, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44.
Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–73.
Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–7.
Shih C, Shilo B, Goldfarb M, Dannenberg A, Weinberg R. Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc Natl Acad Sci U S A. 1979;76(11):5714–8.
Spector D, Varmus H, Bishop J. Nucleotide sequences related to the transforming gene of avian sarcoma virus are present in DNA of uninfected vertebrates. Proc Natl Acad Sci U S A. 1978;75(9):4102–6.
Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13(6):472–82.
Medina M, Sánchez-Jiménez F, Márquez J, Rodríguez Quesada A. Núñez de Castro I. Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem. 1992;113(1):1–15.
Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207–19.
Cheng T, Sudderth J, Yang C, Mullen A, Jin E, Matés J, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;24(108):8674–9.
Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell. 2012;22(5):631–44.
Terunuma A, Putluri N, Mishra P, Mathé E, Dorsey T, Yi M, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124(1):398–412.
Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015;22(6):1068–77.
Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011;8, e1002229.
McCormick F. Ras-related proteins in signal transduction and growth control. Mol Reprod Dev. 1995;42(4):500–6.
Chiaradonna F, Gaglio D, Vanoni M, Alberghina L. Expression of transforming K-Ras oncogene affects mitochondrial function and morphology in mouse fibroblasts. Biochim Biophys Acta. 2006;1757(9-10):1338–56.
Chiaradonna F, Sacco E, Manzoni R, Giorgio M, Vanoni M, Alberghina L. Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene. 2006;25(39):5391–404.
Vizan P, Boros L, Figueras A, Capella G, Mangues R, Bassilian S, et al. K-ras codon-specific mutations produce distinctive metabolic phenotypes in NIH3T3 mice [corrected] fibroblasts. Cancer Res. 2005;65(13):5512–5.
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325(5947):1555–9.
Weinberg F, Hamanaka R, Wheaton W, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107(19):8788–93.
Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523.
Kong B, Qia C, Erkan M, Kleeff J, Michalski CW. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol. 2013;4:246.
Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–32.
Chakrabarti G, Moore ZR, Luo X, Ilcheva M, Ali A, Padanad M, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metab. 2015;3(12). doi:10.1186/s40170-015-0137-1.
Zhang XD, Qin ZH, Wang J. The role of p53 in cell metabolism. Acta Pharmacol Sin. 2010;31(9):1208–12.
Bensaad K, Tsuruta A, Selak M, Vidal M, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–20.
Matoba S, Kang J, Patino W, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–3.
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107(16):7455–60.
Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature. 2013;493(7434):689–93.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107(16):7461–6.
Szeliga M, Obara-Michlewska M, Matyja E, Łazarczyk M, Lobo C, Hilgier W, et al. Transfection with liver-type glutaminase cDNA alters gene expression and reduces survival, migration and proliferation of T98G glioma cells. Glia. 2009;57(9):1014–23.
Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol Cell. 2013;50(2):200–11.
Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23(5):537–48.
Haigis M, Guarente L. Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20(21):2913–21.
Fernandez-Marcos PJ, Serrano M. Sirt4: the glutamine gatekeeper. Cancer Cell. 2013;23(4):427–8.
Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell. 2013;23(4):450–63.
Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153(4):840–54.
Cetinbas N, Daugaard M, Mullen AR, Hajee S, Rotblat B, Lopez A, et al. Loss of the tumor suppressor Hace1 leads to ROS-dependent glutamine addiction. Oncogene. 2014.
Menon S, Manning B. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008; Suppl 2:S43–51.
Bentzinger C, Romanino K, Cloëtta D, Lin S, Mascarenhas J, Oliveri F, et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008;5:411–24.
Elstrom R, Bauer D, Buzzai M, Karnauskas R, Harris M, Plas D, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–9.
Gottlob K, Majewski N, Kennedy S, Kandel E, Robey R, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15(11):1406–18.
Ramanathan A, Schreiber S. Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci U S A. 2009;106(52):22229–32.
Gingras A, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15(7):807–26.
Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47(3):349–58.
Shanware NP, Bray K, Eng CH, Wang F, Follettie M, Myers J, et al. Glutamine deprivation stimulates mTOR-JNK-dependent chemokine secretion. Nat Commun. 2014;5:4900.
Onul A, Elseth K, De Vitto H, Paradise W, Vesper B, Tarjan G, et al. Long-term adaptation of the human lung tumor cell line A549 to increasing concentrations of hydrogen peroxide. Tumour Biol. 2012;33(3):739–48.
Vesper B, Onul A, Haines GR, Tarjan G, Xue J, Elseth K, et al. Part I. Molecular and cellular characterization of high nitric oxide-adapted human breast adenocarcinoma cell lines. Tumour Biol. 2013;24(1):203–14.
Jung C, Ro S, Cao J, Otto N, Kim D. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–95.
Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell. 2015;162(2):259–70.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62.
Davies S, Sim A, Hardie D. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem. 1990;187(1):183–90.
Gwinn D, Shackelford D, Egan D, Mihaylova M, Mery A, Vasquez D, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26.
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41.
Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17(1):113–24.
Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012;483(7391):608–12.
Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell. 2013;152(3):599–611.
Muñoz-Pinedo C, El Mjiyad N, Ricci JE. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012;3, e248.
Ames B. Dietary Carcinogens and Anticarcinogens. Science. 1983;221(4617):1256–64.
De Vitto H, Mendonça B, Elseth K, Onul A, Xue J, Vesper B, et al. Part III. Molecular changes induced by high nitric oxide adaptation in human breast cancer cell line BT-20 (BT-20-HNO): a switch from aerobic to anaerobic metabolism. Tumour Biol. 2013;34(1):403–13.
Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313:17–29.
Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, et al. Cyclic changes in metabolic state during the life of a yeast cell. Proc Natl Acad Sci U S A. 2007;104(23):16886–91.
Dasgupta S, Soudry E, Mukhopadhyay N, Shao C, Yee J, Lam S, et al. Mitochondrial DNA mutations in respiratory complex-I in never-smoker lung cancer patients contribute to lung cancer progression and associated with EGFR gene mutation. J Cell Physiol. 2012;227(6):2451–60.
Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;11:857–66.
King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25(34):4675–82.
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2015;29(8):887.
Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477(7363):225–8.
Rajagopalan KN, Egnatchik RA, Calvaruso MA, Wasti AT, Padanad MS, Boroughs LK, et al. Metabolic plasticity maintains proliferation in pyruvate dehydrogenase deficient cells. Cancer Metab. 2015;3:7.
Chowdhury R, Yeoh K, Tian Y, Hillringhaus L, Bagg E, Rose N, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–9.
Lu C, Ward P, Kapoor G, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–8.
Ward P, Patel J, Wise D, Abdel-Wahab O, Bennett B, Coller H, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–34.
Icard P, Poulain L, Lincet H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim Biophys Acta. 2012;1825(1):111–6.
Hatzivassiliou G, Zhao F, Bauer D, Andreadis C, Shaw A, Dhanak D, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8(4):311–21.
Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491(7424):458–62.
Méndez-Lucas A, Hyroššová P, Novellasdemunt L, Viñals F, Perales JC. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER)stress response gene involved in tumor cell adaptation to nutrient availability. J Biol Chem. 2014;289(32):22090–102.
Montal ED, Dewi R, Bhalla K, Ou L, Hwang BJ, Ropell AE, et al. PEPCK coordinates the regulation of central carbon metabolism to promote cancer cell growth. Mol Cell. 2015;60(4):571–83.
Klarer AC, O’Neal J, Imbert-Fernandez Y, Clem A, Ellis SR, Clark J, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014;2(1):2.
Berg J, Tymoczko J, Stryer L. The glycolytic pathway is tightly controlled. biochemistry. 5th ed. New York, USA: W H Freeman; 2002.
Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;9(4):191–8.
Possemato R, Marks K, Shaul Y, Pacold M, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476(7360):346–50.
Kalhan S, Hanson R. Resurgence of serine: an often neglected but indispensable amino acid. J Biol Chem. 2012;287(24):19786–91.
Locasale J, Grassian A, Melman T, Lyssiotis C, Mattaini K, Bass A, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43(9):869–74.
Maddocks O, Berkers C, Mason S, Zheng L, Blyth K, Gottlieb E, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493(7433):542–6.
Amelio I, Markert E, Rufini A, Antonov A, Sayan B, Tucci P, et al. p73 regulates serine biosynthesis in cancer. Oncogene. 2014;33(42):5039–46.
Duthie S. Folate and cancer: how DNA damage, repair and methylation impact on colon carcinogenesis. J Inherit Metab Dis. 2011;34(1):101–9.
Piskac-Collier A, Monroy C, Lopez M, Cortes A, Etzel C, Greisinger A, et al. Variants in folate pathway genes as modulators of genetic instability and lung cancer risk. Genes Chromosomes Cancer. 2011;50(1):1–12.
Farber S, Diamond L, Mercer R, Sylvester R, Wolff J. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (aminopterin). N Engl J Med. 1948;238:787–93.
Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev. 2010;3(1):23–34.
Holmström K, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–21.
Cooke M, Evans M, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195–214.
Marnett L. Oxyradicals and DNA damage. Carcinogenesis. 2000;21(3):361–70.
Valko M, Rhodes C, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40.
Amores-Sánchez M, Medina M. Glutamine, as a precursor of glutathione, and oxidative stress. Mol Genet Metab. 1999;67(2):100–5.
Roth E, Oehler R, Manhart N, Exner R, Wessner B, Strasser E, et al. Regulative potential of glutamine—relation to glutathione metabolism. Nutrition. 2002;18(3):217–21.
Estrela J, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. 2006;43(2):143–81.
Gañán-Gómez I, Wei Y, Yang H, Boyano-Adánez M, García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med. 2013;65:750–64.
Hayes J, McMahon M. The double-edged sword of Nrf2: subversion of redox homeostasis during the evolution of cancer. Mol Cell. 2006;21(6):732–4.
Kensler T, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31(1):90–9.
Kaspar J, Niture S, Jaiswal A. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47(9):1304–9.
Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22(1):66–79.
Afonyushkin T, Oskolkova OV, Philippova M, Resink TJ, Erne P, Binder BR, et al. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependentmechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler Thromb Vasc Biol. 2010;30(5):1007–13.
He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276(24):20858–65.
DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47(12):1475–81.
Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–47.
Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–62.
Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–98.
DeBerardinis R, Lum J, Hatzivassiliou G, Thompson C. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;1:11–20.
Meng M, Chen S, Lao T, Liang D, Sang N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell Cycle. 2014;9(19):3921–32.
Eng C, Abraham R. Glutaminolysis yields a metabolic by-product that stimulates autophagy. Autophagy. 2010;7:968–70.
Mariño G, Kroemer G. Ammonia: a diffusible factor released by proliferating cells that induces autophagy. Sci Signal. 2010;124:pe19.
Cheong H, Lindsten T, Thompson C. Autophagy and ammonia. Autophagy. 2012;1:122–3.
Green D, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345(6203):1250256.
Weinberg S, Chandel N. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;1:9–15.
Ahluwalia G, Grem J, Hao Z, Cooney D. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther. 1990;46(2):243–71.
Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–21.
Seltzer M, Bennett B, Joshi A, Gao P, Thomas A, Ferraris D, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–7.
Thornburg J, Nelson K, Clem B, Lane A, Arumugam S, Simmons A, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10(5):R84.
Stalnecker C, Ulrich SL Y, Ramachandran S, McBrayer M, DeBerardinis R, Cerione R, et al. Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc Natl Acad Sci U S A. 2015;112(2):394–9.
Narta U, Kanwar S, Azmi W. Pharmacological and clinical evaluation of L-asparaginase in the treatment of leukemia. Crit Rev Oncol Hematol. 2007;61(3):208–21.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–33.
Jin L, Alesi GN, Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. 2015. doi:10.1038/onc.2015.447.
Li B, Simon MC. Molecular pathways: targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin Cancer Res. 2013;1:19(21). doi:10.1158/1078-0432.
Kim JW, Amin AR, Shin DM. Chemoprevention of head and neck cancer with green tea polyphenols. Cancer Prev Res (Phila). 2010;3(8):900–9.
Qu W, Oya S, Lieberman BP, Ploessl K, Wang L, Wise DR, et al. Preparation and characterization of L-[5-11C]-glutamine for metabolic imaging of tumors. J Nucl Med. 2012;53(1):98–105.
Lieberman BP, Ploessl K, Wang L, Qu W, Zha Z, Wise DR, et al. PET imaging of glutaminolysis in tumors by 18F-(2S,4R)4-fluoroglutamine. J Nucl Med. 2011;52(12):1947–55.
Venneti S, Dunphy M, Zhang H, Pitter K, Zanzonico P, Campos C, et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med. 2015;7(274):274ra17.
Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49(3):388–98.
Metallo C, Walther J, Stephanopoulos G. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol. 2009;144(3):167–74.
Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15(2):157–70.
Mayers J, Vander HM. Famine versus feast: understanding the metabolism of tumors in vivo. Trends Biochem Sci. 2015.
Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75(3):544–53.
Acknowledgments
This work was funded by the Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq), Brazil (grant number BEX 9281/13-4).
Author contributions
Study concept and design: Humberto De Vitto; drafting of the manuscript: Humberto De Vitto, Juan Pérez-Valencia, James A. Radosevich; critical revision of the manuscript for intellectual content: Humberto De Vitto, Juan Pérez-Valencia, James A. Radosevich.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Rights and permissions
About this article

Cite this article
De Vitto, H., Pérez-Valencia, J. & Radosevich, J.A. Glutamine at focus: versatile roles in cancer. Tumor Biol. 37, 1541–1558 (2016). https://doi.org/10.1007/s13277-015-4671-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4671-9