
Abstract
Metastasis accounts for more than 50 % of deaths among renal cell carcinoma (RCC) patients, and therefore, it is important to study the biology of metastasis and identify metastasis-associated biomarkers for risk prognosis and stratification of patients for an individualized therapy of RCC. In cultured RCC cells, knockdown of Rictor by short hairpin RNA (shRNA) inhibited cell migration and invasion, probably due to impairments in activation of Akt. Pretreatment with tumor necrosis factor α (TNFα) or interleukin 6 (IL-6) enhanced the expression of Rictor and the migration of renal cancer cells. Mechanistic analysis showed that TNFα induced the activation of NF-κB in RCC cells. Luciferase reporter analysis revealed a NF-κB responding element (−301 to −51 bp) at the promoter region of Rictor. Chromatin immunoprecipitation (ChIP) analysis further confirmed that TNFα-induced binding of p65 with the promoter of Rictor. In a xenograft model, knockdown of Rictor-blocked RCC cells metastasis to the mouse lungs and livers. Taken together, our results suggest that the proinflammatory cytokine TNFα promotes the expression of Rictor through the NF-κB pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Renal cell carcinoma (RCC) accounts for two percent of all cancers [1]. Its incidence has been increasing in the last three decades [2]. More than 30 % of RCC patients are diagnosed as metastatic disease and 20–30 % of patients will finally develop metastases following surgery, immunotherapy, and molecular-targeted therapy. Metastasis is the leading cause of mortality in RCC [3]. Thus, the prevention and treatment of metastasis is of critical importance during RCC therapies.
Chronic inflammation promotes tumorigenesis and is proposed to be a hallmark of cancer [4, 5]. Reducing inflammation is a promising strategy for the prevention of cancers. Clinical investigations reveal a repertoire of proinflammatory cytokines elevated in tumor sites [6, 7]. Among them, TNFα and IL-6 are closely associated with cancer cell metastasis in several types of cancers [8–11]. In RCC, the elevation of TNFα was also detected in the primary sites and linked to a poor prognosis [12]. It has been proposed that TNFα is involved in epithelial-mesenchymal transition and promotes metastasis [8, 13]. However, the role and the mechanism of TNFα-promoted RCC metastasis are still largely unknown.
NF-κB, a transcription factor, plays a pivotal role in a spectrum of biological responses including inflammation and embryonic development [4, 14]. Upon stimulation by extracellular factors such as cytokines, phosphorylation of IKK triggers degradation of IκB which in turn activates downstream transcription [15]. Recently, the role of NF-κB signaling has emerged in cancer development and progression. As the downstream of the TNF pathway, NF-κB provides a mechanistic link between inflammation and tumorigenesis [16–18]. However, the details of the NF-κB pathway in inflammation-related RCC metastasis remain largely unknown.
Metastasis is a multiple-step process which involves escaping of primary tumor cells from the original tissue, intravasating into a blood vessel, traveling through the circulatory system, and finally extravasating to secondary tissues [14]. In breast cancer, epidermal growth factor (EGF)-induced chemotaxis plays a pivotal role in the invasion and metastasis of breast tumors [19]. EGF stimulation results in the activation of PDK1-Akt signaling which, in turn, leads to cofilin-mediated cytoskeleton rearrangement and enhances cell adhesion through integrin [20–22]. Deregulation of the signaling pathways regulated by growth factors and chemokines has been linked to RCC metastasis [23]. However, the underlying molecular mechanism is largely unknown.
The mammalian target of rapamycin (mTOR) plays a pivotal role in tumorigenesis and metastasis [24, 25]. mTOR forms a rapamycin-sensitive mTORC1 complex and Rictor-containing rapamycin insensitive mTORC2 complex [26]. mTORC1 is a central integrator and processor of intracellular and extracellular signals, and controls cell growth, proliferation, survival, and metabolism [26, 27]. mTORC2 promotes the activation of Akt signaling by phosphorylating Akt at Ser473 and regulates cell migration by controlling the dynamics of actin cytoskeletons [28–31]. Targeting mTOR is one of the main current strategies for the development of anticancer drugs, with numerous mTOR kinase inhibitors in preclinical and clinic trials [32–34]. We previously discovered that Rictor is an important mediator of chemotaxis and metastasis in breast cancer cells [35]. In the present study, we investigated the role and mechanism of Rictor in TNFα-promoted RCC migration and metastasis.
Materials and methods
Cell culture
HUVEC, MCF-10A, MDA-MB-231, CRL1932, CRL1933, ACHN, HEK293T cells were obtained from the American Type Culture Collection. MDA-MB-231, CRL1932 and CRL1933 cells were cultured in RPMI-1640. ACHN cells were cultured in Minimum Essential Medium (MEM), while HEK293T cells were grown in Dulbecco's Modified Eagle's medium (DMEM). HUVEC cells were cultured in F-12K Medium supplemented with 100 μg/ml heparin and 50 μg/ml endothelial cell growth supplement (ECGS). MCF-10A cells were cultured in DMEM/F12 medium supplemented with 10 μg/mL insulin, 500 ng/mL hydrocortisone, and 100 ng/mL cholera toxin. All cell lines were grown in corresponding medium supplemented with 10 % (v/v) fetal bovine serum (FBS), penicillin G (100 units/ml) and streptomycin (100 units/ml) in a 5 % CO2-humidified incubator at 37 °C. For TNFα or IL-6 pretreatment, cells in log phase were starved overnight, and then were cultured in the basic medium containing 10 ng/ml TNFα or 20 ng/ml IL-6. After 48 h, cells were harvested for further experiments.
Reagents and antibodies
Recombinant human epidermal growth factor (EGF), bovine fibronectin, IL-6 and TNFα were obtained from R&D systems (Minneapolis, MN, USA). Antibodies against Rictor (#2114S), p-Akt (Ser473, #4060S), p-Akt (Thr308, #4056S), Akt (#9272S), and p-IκBα (#9246S) were obtained from Cell Signaling Technology (Beverly, USA). Antibodies against β-actin (sc-47778), IκBα (sc-371), and p65 (sc-372) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
RNA isolation, reverse transcription-PCR, and real-time PCR
Total RNA from cultured cells were extracted with TRIzol method (Ambion, USA). Reverse transcription and real-time PCR were performed according to the manufacturer’s instructions (Takara, Japan). In real-time PCR, the forward and reward primers for RICTOR were 5′-TTTCGGGGATTTCTGGATG-3′ and 5′-AAA GCCCAGTCTCATGACATT-3′, respectively. And the forward and reward primers for GAPDH were 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′-GAAGATGGTGAT GGGATTTC-3′, respectively.
Lentivirus infection
For production of the lentivirus particles, a shRNA (shRictor-Sense: 5′-CCGGTACTTGTGAAGAATCGTATCTTCTCGAGAAGATACGATTCTTCACAAGTTTTTTG-3′; shRictor-Antisense: 5′-AATTCAAAAAACTTGTGAAGAATCGT ATCTTCTCGAGAAGATACGATTCTTCACAAGTA-3′) expression plasmid and a vector containing a scrambled sequence were inserted into pLKO.1-puro plasmid. 293 T cells were transfected with the package and expression plasmids through LipofectamineTM 2000 system. The culture supernatant was harvested 48 h after transfection and centrifuged at 1000 rpm for 5 min to remove cellular debris. For stable clones, ACHN cells were infected with the lentivirus. 6 hours later, cells culture medium was changed into 10 % FBS-MEM. Five days later, puromycin was added into the culture medium at a final concentration of 20 ng/ml.
Dual-luciferase assay
Total DNA from cultured cells were extracted with a DNA extraction kit (Takara, Japan). The sequence of the homo RICTOR promoter region was assessed through Ensembl database. The putative binding site between transcription factors and promoter region of RICTOR was predicted through TFSEARCH database. The various DNA fragments upstream of the RICTOR promoter were amplified from the DNA of ACHN cells, digested with restriction enzymes and inserted into pGL3-basic (Promega, USA). The common reverse primer for inserts was 5′-AAGCTTATTGA CGGGTTTCAGTC-3′. The forward primers for the fragments of −1051 bp, −401 bp, −301 bp, and −51 bp were 5′-GCTAGCTTCAGTCTCATGGAATAG-3′, 5′-GCTAGCGGGCCGTCTATG-3′, 5′-GCTAGCCTGCATTTGGACGAC-3′, and 5′-TCTAGCTAGCGGCGGGCGCGGCGCGCGGGGA-3′, respectively. 1.5 × 105 ACHN cells per well were planted in a 24-well plate the day before transfection. 800 ng of plasmid DNA, adjusted for insert sizes to provide equal molar plasmids, was transiently transfected into cells with LipofectamineTM 2000. 16 ng of pRL-CMVRenilla luciferase reporter (Promega, USA) was used as reference. After 48 h, cells were harvested, lysed, and assayed for luciferase activity using Dual-Luciferase™ reporter assay system (Promega, USA) following the manufacturer’s instructions.
Chromatin immunoprecipitation assay (ChIP)
Cells were starved overnight in a 10 cm dish and pretreated with TNFα (10 ng/ml) or MEM for only 30 min. Cells were fixed with 1 % formaldehyde at 37 °C for 10 min followed by washing and sonication. Chromatin and 7 μg anti-p65 antibody were subjected to ChIP assay as described previously [36]. Real-time PCR was used to quantitate the immunoprecipitated and input DNA fractions via ABI Prism 7500 system (Applied Biosystems, USA) using α-actin as a negative control while IκBα was used as a positive control (see Table 1 for the sequences of primers).
Western blotting assay
Cells were lysed on ice in a RIPA lysis buffer (Cell Signaling Technology, USA) supplemented with protease inhibitors and a phosphatase inhibitor (Roche, Switzerland), and 1 mM PMSF (Sigma, USA). Total proteins of 30 μg were separated by 8 or 10 % SDS-PAGE and transferred onto PVDF membrane, which was blocked with 5 % nonfat milk followed by incubation with primary antibodies. Signals were developed using enhanced chemiluminescence reagents (Millipore, USA) after incubation with an HRP-conjugated secondary antibody.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as described previously with minor modifications [37]. Briefly, cells were plated in 12-well plates containing sterile coverslips pre-coated with polylysine. After 24 h, cells were starved in a serum-free MEM for at least 3 h. After stimulation with 10 ng/mL TNFα for 15 min at 37 °C, cells were fixed with 4 % paraformaldehyde, permeabilized in 0.2 % Triton X-100 in PBS, and blocked in 3 % bovine serum albumin. Subsequently, anti-p65 antibody, Alexa Fluor 488–conjugated secondary antibody and DAPI were used to stain the cells. Coverslips were mounted in slides and visualized with laser scanning microscopy.
Chemotaxis assay
Chemotaxis assay was performed in a 48-well micro-Boyden chamber (Neuroprobe, USA) as described by Sun et al [37]. Briefly, 30 μl of different concentrations of EGF were added into the lower chamber. The 8 μm fibronectin-coated polycarbonate filter membrane was placed between the chambers. 50 μl of cells were suspended in binding medium (MEM, 0.1 % BSA, and 25 mM HEPES) at the density of 7 × 105 cells/ml and were added into the upper chamber. The chamber was incubated at 37 °C in 5 % CO2 for 3 or 5 h. The membrane was rinsed, fixed, and stained. Migrating cells were enumerated at 400× total magnification by light microscopy.
Invasion assay
Transwell inserts for 24-well plates (Costar) were coated with prediluted Matrigel (1 mg/ml). A total of 1 × 105 cells were loaded into the upper compartment of the chambers. Basic medium containing 1 ng/ml EGF was added into the lower chambers. The cells were allowed to invade through the membrane at 37 °C for 5 h. The non-migrating cells on the upper surface were subsequently removed. The membranes were then fixed and stained. Cells migrating through the membranes in five fields were counted under a microscope at 400× magnification.
Wound healing assay
Cells were plated in 6-well plates and cultured to form a monolayer. After serum starvation for 12 h, a single linear wound was created using a 10 μl pipette tip. Cells were transferred to fresh MEM medium with 0.5 % FBS at 37 °C in 5 % CO2 and the distance of the wounds was measured under a light microscope. All samples were tested in triplicate, and the results were expressed as mean ± SD.
Xenograft tumor transplant mouse model
In vivo metastasis assay was performed as described previously [22]. Briefly, Cells were trypsinized and harvested in log phase, and then washed four times with PBS. 3 × 106 cells were injected subcutaneously into 4-week-old male nu/nu mice (n = 5 per group). 12 weeks later, the mice were sacrificed, and the tumors were isolated, and then the lungs and livers were fixed with formalin and embedded in paraffin. Serial sections and H&E staining were performed to detect micrometastasis. Tumors were excised and measured by largest (a) and smallest (b) diameters to calculate tumor volume by V = ab 2/2.
Statistical analysis
The significance in animal metastasis assay was assessed using Pearson chi-square test. Data analysis was performed using SPSS version 17.0 (Chicago, IL, USA). A P value of <0.05 was considered statistically significant. In chemotaxis, migration, and invasion assay, the results were representative of at least three independent experiments and were expressed as the mean ± SD. Different values between groups were compared using t test in GraphPad Prism 5. A P value of <0.05 was considered statistically significant.
Results
Knockdown of Rictor impaired chemotaxis and invasion of RCC cells
First, we detected the expression of Rictor by Western blotting analysis in two normal cell lines and four cancer cell lines. As shown in Fig. 1a, two metastatic human cancer cell lines, MDA-MB-231 and ACHN, expressed high levels of Rictor protein while its expression in two normal cell lines, HUVEC and MCF-10A, or in two primary RCC cell lines, CRL1932 and CRL1933, was much lower, suggesting a role of Rictor in metastasis. Chemotaxis plays a critical role in metastasis. To reveal the role of Rictor in renal cell chemotaxis, a stable clone, designated as shRictor, was established through a lentivirus system. Compared to normal ACHN cells and control cells (designated as Scr), over 80 % of Rictor was reduced both in the protein and mRNA level (Fig. 1b). Wound healing assays, performed in a medium with 0.5 % fetal bovine serum to exclude the effects of cell proliferation, further confirmed the migration defect in shRictor cells (Fig. 1c). In matrigel assay, shRictor cells showed approximately 60 % decrease in invasiveness (Fig. 1d). Consequently, EGF-induced chemotaxis of ACHN cells was significantly impaired as indicated by micro-Boyden chamber assays (Fig. 1e). Western blotting further verified that Rictor knockdown abolished the EGF-induced Akt phosphorylation at Ser473 (Fig. 1f). These results suggest that Rictor is required for renal cancer cell migration and invasion.

Knockdown Rictor inhibited RCC migration. a Western blotting was performed to detect Rictor expression in normal cells, primary, and metastatic cancer cell lines. b Left panel: Real-time PCR demonstrated the expression of Rictor in shRictor stable clone. Right panel: Western blotting analysis demonstrated the expression of Rictor in shRictor stable clone with β-actin as a loading control. c Wound healing assay of ACHN, Scr and shRictor cells. The gap distance on cell monolayer was measured at 0, 3, 6, 9, 12, and 24 h after scratch. The images were photographed at 0 and 24 hours (100× total magnification). d EGF-induced cell invasion was decreased in shRictor cells. e EGF-induced cell chemotaxis was decreased in shRictor cells. f Western blotting analysis of EGF-induced phosphorylation of Akt T308 and S473 in total cell lysate from Scrambled and shRictor cells. Total Akt was used as a loading control
TNFα and IL-6 enhanced Rictor expression and promote cell migration
It has been documented that the expression of TNFα is associated with a poor prognosis of RCC [12]. We next tested its role in mediating RCC migration. As shown in Fig. 2a, treatment with TNFα enhanced the expression of Rictor. Another proinflammatory cytokine, IL-6, also elevated Rictor expression. In chemotaxis assay, pretreatment of TNFα or IL-6 could increase the EGF-induced RCC chemotaxis, nevertheless, the knockdown of Rictor abolished this effect (Fig. 2b). Next, in wound healing assays, treatment with TNFα or IL-6 significantly promoted the migration of RCC cells, while the knockdown of Rictor attenuated TNFα or IL-6 induced enhancement in cell migration (Fig. 2c). Finally, in the matrigel assays, RCC cells showed accelerated migration in the presence of TNFα or IL-6. The knockdown of Rictor blocked the effect (Fig. 2d). Taken together, these results indicate that TNFα or IL-6 increases cell chemotaxis, migration and invasion, all of which were at least partially declined by the knockdown of Rictor.

TNFα or IL-6 increased cell migration through upregulation of the Rictor expression a ACHN cells were stimulated by TNFα or IL-6 for 48 h. Western blotting showed the Rictor expression of ACHN cells pretreated with TNFα or IL-6 in different concentrations. b TNFα or IL-6 pretreatment could enhance EGF-induced cell chemotaxis, while Rictor knockdown impaired the augmentation posed by TNFα or IL-6 pretreatment in cell chemotaxis assay. c TNFα or IL-6 pretreatment could enhance cell migration, which was abolished by Rictor knockdown. d TNFα or IL-6 pretreatment could enhance cell invasion, while Rictor knockdown impaired this effect
TNFα stimulation increased Rictor expression in renal cancer cells through NF-κB pathway
We next determined the downstream signaling of TNFα that regulates the gene transcription of RICTOR. NF-κB is one of the most important downstream molecules in TNFα signaling [38–41]. Re-distribution of p65 from cytosol to nucleus is the former of NF-κB activation. Fluorescence microscopy showed that p65 mainly localized in cytosol (Fig. 3a), treatment with TNFα enhanced its nuclear distribution, suggesting TNFα could activate NF-κB in RCC cells. In response to TNFα, phosphorylation and degradation of IκBα, the inhibitory binding protein of NF-κB, results in activation of NF-κB pathway. Indeed, treatment of ACHN cells with TNFα for 5 min led to the apparent phosphorylation of IκBα, accompanied with a decrease of total IκBα (Fig. 3b).

TNFα induced the gene expression of RICTOR through the NF-κB pathway a ACHN cells were treated with 10 ng/ml TNFα for 15 min. In immunofluorescence assay, TNFα could induce p65 translocation from cytosol to nuclear. b ACHN cells were treated with 10 ng/ml TNFα for the time as indicated. Immunoblots were performed to determine the phosphorylation and total level of IκBα c Dual-luciferase assay illustrated that the luciferase activity was apparently decreased following depletion of the predictive NF-κB binding sites (from residues −301 bp to −51 bp) in RICTOR promoter region. However, the absence of other predictive transcription factors (SP1 and AP2) did not affect the luc activity negatively. d Five pair primers were designed according to the RICTOR promoter sequence. ChIP was performed using an antibody against p65. Immunoprecipitated DNA was analyzed by real-time PCR. The results indicated that p65 was enriched in predictive NF-κB binding sites, and TNFα could promote p65 enrichment. e Bay 11-7082, a NF-κB inhibitor, inhibited TNFα or IL-6 induced Rictor expression
To investigate the transcriptional regulation of RICTOR, four fragments of the RICTOR promoter region were cloned into pGL3-basic plasmid on the basis of transcription prediction. Luciferase assay demonstrated that the fragment from −51 bp to −301 bp, which contained two predictive NF-κB binding sites, was vital to the promoter activity of RICTOR gene (Fig. 3c). To further test our hypothesis that TNFα stimulates the gene transcription of RICTOR via NF-κB signaling, we designed five pairs of primers for ChIP assay and found that primers designed for predictive NF-κB binding site showed about three times affinity than control IgG (Fig. 3d). Most importantly, TNFα increased the binding of p65 to predictive NF-κB binding site of the RICTOR promoter region (Fig. 3d). Subsequently, Bay 11-7082, an inhibitor of IκBα phosphorylation, blocked the IκBα degradation, which also blocked the TNFα and IL-6 induced expression of Rictor (Fig. 3e). These results indicate that TNFα stimulation increases RICTOR expression in renal cancer cells through NF-κB pathway.
Knockdown of Rictor inhibited tumor growth and metastasis in vivo
To extend our observations in vivo, we subcutaneously implanted ACHN cells with lentivirus expressing control (Scr group) or Rictor (shRictor group) short hairpin RNA (shRNA) to nu/nu mice. After 12 weeks, a significant reduction of primary tumor size was observed in the shRictor group, indicating that Rictor played an important role in the growth of RCC (Fig. 4a, b). Furthermore, 80 % of Scr mouse xenografts showed tumor metastasis to lung or liver (Fig. 4c, 4d, e, f), whereas no apparent metastasis sites were detected in shRictor tumor xenografts after 12 weeks. Taken together, our animal experiments indicate that Rictor is required for RCC growth and metastasis in vivo.

Stable silencing of Rictor reduced cancer cell growth and metastasis in vivo a Comparison of tumor size in nu/nu mice implanted with Scr or shRictor cells. b Comparison of tumor volume in nu/nu mice implanted with Scr or shRictor cells. c Comparison of representative lung and liver metastasis in nu/nu mice implanted with Scr or shRictor cells. The arrow pointed to lung tissue, which was not filling with Indian ink due to the bronchus blockade caused by the lung metastasis. d Comparison of human tumor foci on mouse lungs, which were visualized by H&E staining. e Comparison of human tumor foci on mouse livers, which were visualized by H&E staining. f Comparison of the quantity of tumor metastasis cases between Scr and shRictor groups
Discussion
In the present study, we have revealed a new mechanism by which inflammation promoted RCC tumorigenesis and metastasis. Chronic inflammation has been shown to be closely associated with RCC [8, 12]. TNFα, a proinflammatory cytokine, induced epithelial-mesenchymal transition and promoted metastasis and resistance to the sunitinib [12]. IL-6 stimulated tumor growth and angiogenesis via STAT3 [42, 43]. Our results clearly indicated that Rictor was a downstream effector of both TNFα and IL-6. Treatment with TNFα activated NF-κB and promoted Rictor expression. TNFα-induced cell migration was reversed by knocking down Rictor. A NF-κB responsive region, located at RICTOR promoter region, was identified by both luciferase reporter assays and ChIP analysis. Animal experiments indicated that Rictor knockdown cells failed to metastasize to the mouse livers and lungs. Thus, our results suggest that Rictor plays an important role in mediating inflammation-promoted RCC growth and metastasis.
The outcome of patients with metastatic RCC is very poor, with a median survival time of 10 months and a 5-year survival rate of <10 % [44]. Immunotherapy including IL-2 and IFN-α, and molecular-targeted agents, such as sorafenib, sunitinib, and temsirolimus, are the current therapy choices for metastatic RCC [45, 46]. However, only a small subgroup of RCC patients responded to these treatment modules and most responsive patients will finally develop a resistance to these drugs sooner or later [47]. Thus, novel molecular targets are needed for the development of effective anti-RCC therapies. Rictor appears to be the convergence of several oncogenic pathways [48, 49]. Proinflammatory cytokines, TNFα and IL-6, promote its expression. Epidermal growth factor mediates RCC chemotaxis via Rictor. Furthermore, Rictor expression is closely associated with tumorigenesis and metastasis. These data indicate that targeting Rictor-mediated signaling pathways is a promising strategy for the treatment of metastatic cancers.
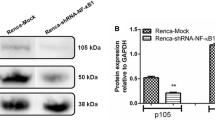
A link between chronic inflammation and cancer has been suspected for more than one century [50]. For example, hepatitis C infection in the liver predisposes to liver, chronic Helicobacter pylori infection with stomach cancer, chronic ulcerative colitis with colon cancer, and so on [51–55]. However, the concept that chronic inflammation has been involved in RCC progression was widely accepted until more recent times [56]. NF-κB, a vital transcription factor, plays a key role in cell apoptosis, proliferation, differentiation and immune response, and is an important molecule in inflammation. As one of the most extensively expressed transcription factors, NF-κB is over activated or upregulated in order to overcome apoptosis and promote cancer cell growth during cancer progression [57, 58]. Furthermore, it has been documented that NF-κB may be involved in caner metastasis [59]. However, the details are largely unknown. In the present study, as the downstream molecule of the proinflammation pathway, NF-κB can bind to the promoter region of RICTOR, and enhance cancer cell migration, chemotaxis and invasion via upregulating Rictor expression. Moreover, Bay 11-7082, an inhibitor of IκBα phosphorylation, could inhibit TNFα and IL-6 induced Rictor upregulation. Taken together, these results demonstrated that a proinflammation cytokine could affect cancer cell metastasis, and the inhibition of the NF-κB pathway may help attenuate inflammation-related metastasis. TNF-α/IL-6/ NF-κB /Rictor axis maybe play a pivotal role in RCC metastasis.
Conclusions
In summary, this study showed that the knockdown of Rictor significantly decreased the metastasis of the RCC cell line ACHN in vitro and in vivo. In addition, proinflammatory cytokines (TNFα and IL-6) might increase RCC metastasis through the upregualtion of the RICTOR transcription. Rictor-mediated signaling is controlled by TNFα NF-κB signaling and EGF-RTK pathway (Fig. 5). The association of the upregulation of Rictor and RCC metastasis suggests that Rictor is a potential biomarker for prognosis and stratification of RCC patents, and molecular target for the development of novel drugs for the treatment of cancer metastasis.

TNFα promotes renal cancer cell metastasis through upregulating the Rictor expression via the NF-κB pathway
References
Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865–75.
Chow WH, Dong LM, Devesa SS. Epidemiology and risk factors for kidney cancer. Nat Rev Urol. 2010;7:245–57.
Wysocki PJ. mtor in renal cell cancer: modulator of tumor biology and therapeutic target. Expert Rev Mol Diagn. 2009;9:231–41.
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–81.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Multhoff G, Molls M, Radons J. Chronic inflammation in cancer development. Front Immunol. 2011;2:98.
Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185.
Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY, Sun GH, et al. TNF-α induces epithelial-mesenchymal transition of renal cell carcinoma cells via a gsk3beta-dependent mechanism. Mol Cancer Res. 2012;10:1109–19.
Hamaguchi T, Wakabayashi H, Matsumine A, Sudo A, Uchida A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem Biophys Res Commun. 2011;407:525–30.
Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the jak-stat3-snail signaling pathway. Mol Cancer Res. 2011;9:1658–67.
Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, et al. The IL-6/JAK/stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15:848–62.
Mikami S, Mizuno R, Kosaka T, Saya H, Oya M, Okada Y. Expression of TNF-α and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int J Cancer. 2015;136:1504–14.
Jung HY, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res. 2015;21:962–8.
Wittekind C, Neid M. Cancer invasion and metastasis. Oncology. 2005;69 Suppl 1:14–6.
Hayden MS, Ghosh S. Signaling to nf-kappab. Genes Dev. 2004;18:2195–224.
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappab functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6.
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. Ikkβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96.
Mauad TH, van Nieuwkerk CM, Dingemans KP, Smit JJ, Schinkel AH, Notenboom RG, et al. Mice with homozygous disruption of the mdr2 p-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol. 1994;145:1237–45.
Xue C, Wyckoff J, Liang F, Sidani M, Violini S, Tsai KL, et al. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res. 2006;66:192–7.
Zhang B, Gu F, She C, Guo H, Li W, Niu R, et al. Reduction of Akt2 inhibits migration and invasion of glioma cells. Int J Cancer. 2009;125:585–95.
Guo H, Gu F, Li W, Zhang B, Niu R, Fu L, et al. Reduction of protein kinase C ζ inhibits migration and invasion of human glioblastoma cells. J Neurochem. 2009;109:203–13.
Liu Y, Wang J, Wu M, Wan W, Sun R, Yang D, et al. Down-regulation of 3-phosphoinositide-dependent protein kinase-1 levels inhibits migration and experimental metastasis of human breast cancer cells. Mol Cancer Res. 2009;7:944–54.
Figlin RA, Kaufmann I, Brechbiel J. Targeting PI3k and mTORC2 in metastatic renal cell carcinoma: new strategies for overcoming resistance to VEGFR and mTORC1 inhibitors. Int J Cancer. 2013;133:788–96.
Polivka Jr J, Janku F. Molecular targets for cancer therapy in the PI3K/Akt/mTOR pathway. Pharmacol Ther. 2014;142:164–75.
Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. 2014;5:49–66.
Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22.
Wullschleger S, Loewith R, Hall MN. Tor signaling in growth and metabolism. Cell. 2006;124:471–84.
Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101.
Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 s6 kinase and 4E-BP1. Proc Natl Acad Sci U S A. 1998;95:1432–7.
Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302.
Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian tor complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8.
Janes MR, Vu C, Mallya S, Shieh MP, Limon JJ, Li LS, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2013;27:586–94.
Bhagwat SV, Gokhale PC, Crew AP, Cooke A, Yao Y, Mantis C, et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther. 2011;10:1394–406.
Naing A, Aghajanian C, Raymond E, Olmos D, Schwartz G, Oelmann E, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br J Cancer. 2012;107:1093–9.
Zhang F, Zhang X, Li M, Chen P, Zhang B, Guo H, et al. mTOR complex component Rictor interacts with PKCζ and regulates cancer cell metastasis. Cancer Res. 2010;70:9360–70.
Liu Z, Garrard WT. Long-range interactions between three transcriptional enhancers, active Vκ gene promoters, and a 3′ boundary sequence spanning 46 kilobases. Mol Cell Biol. 2005;25:3220–31.
Sun R, Gao P, Chen L, Ma D, Wang J, Oppenheim JJ, et al. Protein kinase C ζ is required for epidermal growth factor-induced chemotaxis of human breast cancer cells. Cancer Res. 2005;65:1433–41.
Heyninck K, Beyaert R. Crosstalk between NF-κB-activating and apoptosis-inducing proteins of the TNF-receptor complex. Mol Cell Biol Res Commun. 2001;4:259–65.
Wu Y, Zhou BP. TNF-α/NF-κB/snail pathway in cancer cell migration and invasion. Br J Cancer. 2010;102:639–44.
Li D, Zhong Y, Zhou Y, Sun H, Zheng X, Zhao C, et al. Autocrine TNF-α-mediated NF-κB activation is a determinant for evasion of CD40-induced cytotoxicity in cancer cells. Biochem Biophys Res Commun. 2013;436:467–72.
Prabhavathy D, Subramanian CK, Karunagaran D. Re-expression of HPV16 E2 in SiHa (human cervical cancer) cells potentiates NF-κB activation induced by TNF-α concurrently increasing senescence and survival. Biosci Rep. 2015;35. doi:10.1042/BSR20140160.
De Simone V, Franze E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D, et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-κB to promote colorectal cancer cell growth. Oncogene. 2015;34:3493–503.
Lin CM, Shyu KG, Wang BW, Chang H, Chen YH, Chiu JH. Chrysin suppresses IL-6-induced angiogenesis via down-regulation of JAK1/STATt3 and VEGF: an in vitro and in ovo approach. J Agric Food Chem. 2010;58:7082–7.
Shinohara N, Nonomura K, Abe T, Maruyama S, Kamai T, Takahashi M, et al. A new prognostic classification for overall survival in Asian patients with previously untreated metastatic renal cell carcinoma. Cancer Sci. 2012;103:1695–700.
Escudier B, Albiges L, Sonpavde G. Optimal management of metastatic renal cell carcinoma: current status. Drugs. 2013;73:427–38.
Albouy B, Gross Goupil M, Escudier B, Massard C. Renal cell carcinoma management and therapies in 2010. Bull Cancer. 2010;97:17–28.
Legramanti S, Antonelli A, Ferrari V, Arrighi N, Corti S, Zanotelli T, et al. Simeone C: [results of targeted therapies for m1 renal cell carcinoma: our experience]. Urologia. 2012;79 Suppl 19:72–5.
Bashir T, Cloninger C, Artinian N, Anderson L, Bernath A, Holmes B, et al. Conditional astroglial Rictor overexpression induces malignant glioma in mice. PLoS One. 2012;7:e47741.
Bera A, Das F, Ghosh-Choudhury N, Kasinath BS, Abboud HE, Choudhury GG. Microrna-21-induced dissociation of PDCD4 from Rictor contributes to Akt-IKKβ-mTORC1 axis to regulate renal cancer cell invasion. Exp Cell Res. 2014;328:99–117.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45.
Granado-Serrano AB, Martin MA, Bravo L, Goya L, Ramos S. Quercetin attenuates TNF-induced inflammation in hepatic cells by inhibiting the NF-κB pathway. Nutr Cancer. 2012;64:588–98.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7.
Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STATt-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089–97.
Katsha A, Soutto M, Sehdev V, Peng D, Washington MK, Piazuelo MB, et al. Aurora kinase a promotes inflammation and tumorigenesis in mice and human gastric neoplasia. Gastroenterology 2013;145:1312-1322 e1311-1318.
van der Woude CJ, Moshage H, Homan M, Kleibeuker JH, Jansen PL, van Dekken H. Expression of apoptosis related proteins during malignant progression in chronic ulcerative colitis. J Clin Pathol. 2005;58:811–4.
de Vivar Chevez AR, Finke J, Bukowski R. The role of inflammation in kidney cancer. Adv Exp Med Biol. 2014;816:197–234.
Lu Y, Zhao X, Luo G, Shen G, Li K, Ren G, et al. Thioredoxin-like protein 2b facilitates colon cancer cell proliferation and inhibits apoptosis via NF-κB pathway. Cancer Lett. 2015;363:119–26.
Luo Y, Wang SX, Zhou ZQ, Wang Z, Zhang YG, Zhang Y, et al. Apoptotic effect of genistein on human colon cancer cells via inhibiting the nuclear factor-kappa B (NF-κB) pathway. Tumour Biol. 2014;35:11483–8.
Xia J, Wang F, Wang L, Fan Q. Elevated serine protease HTRA1 inhibits cell proliferation, reduces invasion, and induces apoptosis in esophageal squamous cell carcinoma by blocking the nuclear factor-κb signaling pathway. Tumour biol. 2013;34:317–28.
Acknowledgments
This work was supported by the NFSC (81472683, 81072160, 81201646, and 81125019), 973 Program (2011CB933100), Program of Tianjin Higher School Innovation Team (TD12-5025), and Research Seed Foundation of Tianjin Medical University Cancer Hospital and Institute (1421). We thank Zhe Liu, Wei Du, and Litao Qin for their help in luciferase and ChIP assays, and Qiuping Dong for plasmid construction.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The manuscript does not contain clinical studies or patient data. And all procedures performed in studies involving animals were in accordance with the ethical standards of the ethics committee in Cancer Institute and Hospital of Tianjin Medical University, which was draw up on the basis of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize suffering.
Conflicts of interest
None.
Additional information
Bo Sun and Liwei Chen contributed equally to this work.
Rights and permissions
About this article

Cite this article
Sun, B., Chen, L., Fu, H. et al. Upregulation of RICTOR gene transcription by the proinflammatory cytokines through NF-κB pathway contributes to the metastasis of renal cell carcinoma. Tumor Biol. 37, 4457–4466 (2016). https://doi.org/10.1007/s13277-015-4296-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4296-z