
Abstract
Recombinant human arginase (rhArg) is an arginine-degrading enzyme that has been evaluated as effective therapeutics for varieties of malignant tumors and is in clinical trials for hepatocellular carcinoma (HCC) treatment nowadays. Our previous studies have reported that rhArg could induce autophagy and apoptosis in lymphoma cells and inhibiting autophagy could enhance the efficacy of rhArg on lymphoma. However, whether rhArg could induce autophagy and what roles autophagy plays in leukemia cells are unclear. In this study, we demonstrated that rhArg treatment could lead to the formation of autophagosomes and the upregulation of microtubule-associated protein light chain 3 II (LC3-II) in human promyelocytic leukemia HL-60 cells and human acute T cell leukemia Jurkat cells. Furthermore, inhibiting autophagy using 3-methyladenine (3-MA) or chloroquine (CQ) could significantly enhance rhArg-induced cell growth inhibition and apoptosis. Taken together, these findings indicated that rhArg induced autophagy in leukemia cells and inhibiting autophagy enhanced anti-leukemia effect of rhArg, which might encourage the treatment of leukemia by targeting arginine depletion and autophagy in clinics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leukemia is one of the most death-leading cancers [1]. Thanks to the unremitting efforts in both academic and industrial worlds, a great number of anti-leukemia drugs have been used in clinics for the treatment of different subtypes of leukemia [2, 3]. Although the first-line anti-leukemia drug imatinib mesylate has rewritten the history of leukemia treatment, we have been facing many challenges in providing better treatment and high-quality life for patients with leukemia [4]. These challenges include continuous drug application, high cost, intolerable side effects, and risk of drug resistance [5]. So, it is urgent to develop new strategies for leukemia treatment. Recently, many lines of evidence have shown that amino acid depletion could lead to cell death of leukemia cells [6, 7]. Arginase is an enzyme which can deplete arginine, a semi-essential amino acid, for adult mammals, in cell division, ammonia metabolism, and immune function [8]. Recombinant human arginase (rhArg) has been proved very effective for certain types of cancers such as malignant melanoma, lymphoma, and hepatocellular carcinoma (HCC) and now is in ongoing clinical trial for treatment, but whether rhArg has therapeutic effects on leukemia is not well evaluated [9–11].
Deprivation of nutrients can induce autophagy, which will degrade misfolded proteins or organelles to provide energy for cell survival [12]. Under this circumstance, autophagy tends to play a cytoprotective role and serves as an important factor which could reduce a tumor’s sensitivity to drugs [13]. Increasing evidence demonstrated that autophagy is a drug-resistant mechanism in many types of cancers and inhibition of autophagy could significantly enhance the anti-cancer efficacy of many drugs including imatinib and interferon beta in certain cancers [2, 14]. Our previous study has demonstrated that rhArg could induce autophagy in non-Hodgkin’s lymphoma, and inhibiting autophagy could significantly enhance the anti-lymphoma effect of rhArg [11]. These findings suggested that inhibiting autophagy might be an effective strategy to promote the clinical outcomes of rhArg treatment.
The primary aims of this study were to assess the therapeutic effects of rhArg on leukemia cells, explore the role of autophagy induced by rhArg, and provide new strategies to encourage the therapeutic effects of rhArg by regulating autophagy. In the current work, the therapeutic effects of rhArg on leukemia cells were confirmed by detecting cell growth inhibition and cell cycle distribution. Meanwhile, the formation of autophagosomes and changes of the microtubule-associated protein light chain 3 were also detected to ensure autophagy induced by rhArg. To further investigate whether rhArg-induced autophagy was responsible for the treatment of leukemia, the therapeutic effects of rhArg combined with autophagy inhibitors, 3-methyladenine and chloroquine, were examined. This is the first study to show that autophagy played an antagonism role in rhArg-induced cell death and inhibition of autophagy might be an effective strategy to improve the anti-cancer efficacy of rhArg.
Materials and methods
Cell lines
HL-60 (human promyelocytic leukemia cells) and Jurkat (human acute T cell leukemia cells) were obtained from Cell Bank of the Chinese Academy of Sciences, Shanghai Branch (Shanghai, China). All cells were maintained in RPMI-1640 medium (Invitrogen, San Diego, CA, USA) supplemented with 10 % heat-inactivated fetal bovine serum (Invitrogen, San Diego, CA, USA), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified incubator with 5 % CO2 and 95 % air at 37 °C.
Reagents
rhArg was expressed and purified as previously described [15]. The specific activity of rhArg was approximately 200 IU/ml, and the purity of the recombinant protein is above 95 %. One international unit of rhArg is defined as the amount of enzyme that produces 1 μmol urea/min at 30 °C, pH 8.5.
The phosphatidylinositol 3-phosphate kinase (PI3K) inhibitor 3-methyladenine (3-MA) and the lysosomal inhibitor chloroquine (CQ) were obtained from Sigma (St. Louis, MO, USA). Antibodies to LC3 and β-actin were obtained from Cell Signaling Technology (Danvers, MA, USA). The secondary antibodies horseradish peroxidase (HRP)-conjugated goat anti-mouse and anti-rabbit immunoglobulin G (IgG) were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Annexin V-FITC Apoptosis Detection Kit was purchased from BD Biosciences (Franklin Lakes, NJ, USA). A cell cycle analysis kit was obtained from Beyotime Institute of Biotechnology (Haimen, China).
Cell proliferation assay
Cell proliferation was measured using the MTT assay, as previously described [6]. Briefly, about 10,000 cells per well were seeded in 96-well plates and then exposed to rhArg with or without autophagy inhibitors in indicated concentrations. The cells were incubated with MTT solution (0.5 mg/ml) for 4 h at 37 °C. Then, 100 μl of 20 % SDS in dimethyl formamide/H2O (1:1, v/v; pH 4.7) was added to each well. Evenly shaking the 96-well plates resulted in complete dissolution, and the optical density (OD) was measured at an absorbance wavelength of 570 nm after addition of formazan.
Cell cycle analysis
HL-60 and Jurkat cells were treated with rhArg at indicated concentrations for 48 h. Cells were harvested and stained with PI according to the manufacturer’s instruction. Cell cycle distribution was detected using a flow cytometer (Becton–Dickinson, Fullerton, CA).
Apoptosis assay
Apoptosis was measured using the Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s guides. Analysis was performed using a FACSCalibur flow cytometer (Becton–Dickinson, Fullerton, CA).
Western blot analysis
Western blot analysis was performed as previously described [16]. Briefly, after HL-60 and Jurkat cells were treated with rhArg for 24 h, cells were harvested and an equivalent amount of cell lysates was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred for western blot assay. The polyvinylidene fluoride (PVDF) membranes were incubated with primary antibodies and secondary antibodies and then analyzed by an enhanced chemiluminescent detection kit (Pierce, Rockford, IL, USA).
Transmission electron microscopy
HL-60 and Jurkat cells were incubated with 1 IU/ml of rhArg for 24 h, and then cells were harvested and managed as described [16]. Samples were analyzed with a JEM 1230 transmission electron microscope (JEOL, Japan). Micrographs were taken at ×5000 or ×20,000 magnification.
Confocal fluorescence assay
HL-60 and Jurkat cells were incubated with 1 IU/ml of rhArg for 24 h, and then cells were harvested and disposed with Cyto-ID Autophagy Detection Kit (ENZO Life Science, Farmingdale, NY, USA) following the manufacturer’s instruction. The images were observed by inverted confocal microscopy (Carl Zeiss LSM710, Carl Zeiss, Germany).
Statistics analysis
Statistics analysis was carried out with GraphPad Prism 5. The results were expressed as means ± standard deviations (SD). Comparisons were performed using Student’s t test (two-tailed) or one-way ANOVA. A p value <0.05 was considered statistically significant.
Results
rhArg inhibited the growth of HL-60 and Jurkat cells in vitro
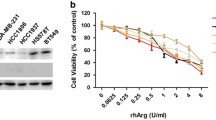
Firstly, we evaluated whether rhArg could inhibit the growth of leukemia cells. HL-60 and Jurkat cells were treated with 0.016, 0.031, 0.063, 0.125, 0.25, 0.5, 1, 2, and 4 IU/ml of rhArg for 48 h, and then cell viability was measured using MTT assay. As shown in Fig. 1a, b, rhArg could significantly inhibit the growth of HL-60 and Jurkat cells in a dose-dependent manner. The cell growth inhibition rate was as high as 73.2 % (p < 0.01) in HL-60 cells and 60.5 % (p < 0.01) in Jurkat cells when rhArg was at the concentration of 4 IU/ml for 48 h. These results confirmed the efficacy of rhArg on leukemia cells and targeting arginine depletion might be a potential therapeutic strategy for leukemia.

rhArg inhibited the growth of HL-60 and Jurkat cells in vitro. a HL-60 cells (1 × 104 cells/well) were incubated for 48 h in the presence or absence of different concentrations of rhArg; cell growth inhibitory effects were analyzed by MTT. The data are presented as the means ± SD of four samples. *p < 0.05 versus control, **p < 0.01 versus control. A typical result from three independent experiments is presented. b Jurkat cells (1 × 104 cells/well) were incubated for 48 h in the presence or absence of different concentrations of rhArg; cell growth inhibitory effects were analyzed by MTT. The data are presented as the means ± SD of four samples. *p < 0.05 versus control, **p < 0.01 versus control. A typical result from three independent experiments is presented
rhArg made significant influences on the cell cycle distribution of leukemia cells
To explore the mechanisms of rhArg for the treatment of leukemia, the effects of rhArg on cell cycle distribution in HL-60 and Jurkat cells were detected. As shown in Fig. 2b, compared with the control group, the proportion of HL-60 cells in the G0/G1 phase was notably increased from 44.0 to 49.0 % and 55.5 % when cells were treated with 0.5 and 1 IU/ml of rhArg, respectively. Meanwhile, the percentage of G2 phase cells was significantly decreased from 15.5 to 7.2 % and 9.5 %, respectively. Moreover, the percentage of cells in the S phase was significantly decreased from 40.5 to 35.0 % when HL-60 cells were treated with 1 IU/ml of rhArg. Similar results were also shown in Jurkat cells (Fig. 2c, d). These results suggested that rhArg could induce cell cycle arrest in the G0/G1 phase in HL-60 and Jurkat cells.

rhArg made significant influences on the cell cycle distribution of leukemia cells. a HL-60 cells (1 × 104 cells/well) were incubated in the presence or absence of different concentrations of rhArg (0.25, 0.5, and 1 IU/ml) for 48 h; cells were stained with PI and analyzed by flow cytometry. The percentages of cells in the G0/G1 phase, G2 phase, and S phase are shown in the bar charts (b). *p < 0.05 versus control, **p < 0.01 versus control. c Jurkat cells (1 × 104 cells/well) were incubated in the presence or absence of different concentrations of rhArg (0.25, 0.5, and 1 IU/ml) for 48 h; cells were stained with PI and analyzed by flow cytometry. The percentages of cells in the G0/G1 phase, G2 phase, and S phase are shown in the bar charts (d). *p < 0.05 versus control, **p < 0.01 versus control
rhArg induced autophagy in leukemia cells
In this study, we evaluated whether rhArg induced autophagy in leukemia cells. Firstly, we used transmission electron microscopy to observe if there was autophagosome formation in rhArg-treated cells, which is a golden standard of autophagy detection. The result showed that there were numerous vacuolizations and electron-dense inclusions accumulated after cells were treated with rhArg for 24 h, compared with untreated cells (Supplementary Fig. 1). Upon magnification, these vacuolizations presented obviously double-layered membrane structures, which appeared to be autophagosomes (Fig. 3a).

rhArg induced autophagy in leukemia cells. a rhArg induced formation of autophagosomes. HL-60 and Jurkat cells were treated with or without 1 IU/ml of rhArg for 24 h. Cell samples were prepared for transmission electron microscopy analysis as described in “Materials and methods” (scale bar = 2 μm). A magnified view of the electron photomicrograph shows a characteristic autophagosome (scale bar = 0.5 μm). b rhArg induced the accumulation of LC3-II. HL-60 and Jurkat cells were treated with 0, 0.1, 0.2, 0.5, and 1 IU/ml of rhArg for 24 h. Cell lysates were analyzed by western blot. Beta-action was used as a protein-loading control. c rhArg induced the accumulation of autophagic fluorescence. HL-60 and Jurkat cells were treated as described in a. Cell samples were prepared for confocal microscopy analysis as described in “Materials and methods.” Untreated cells were used as negative control (scale bar = 10 μm)
In addition, the expression of the microtubule-associated protein 1 light chain 3 (LC3-I) and its membrane-bound form LC3-II was analyzed by western blot. Western blot analysis showed the increase of LC3-II in cells treated with series concentrations of rhArg for 24 h. As LC3-II is highly expressed on the membranes of autophagosomes, these results confirmed that rhArg could induce autophagy in HL-60 and Jurkat leukemia cells (Fig. 3b).
Furthermore, fluorescence microscopy was also used to detect autophagy triggered by rhArg in HL-60 and Jurkat cells. Cyto-ID green dye is a specific marker for autophagic vacuoles [17]. As shown in Fig. 3c, a green fluorescence was detected in the cytoplasm of rhArg-treated and positive control rapamycin-treated HL-60 and Jurkat cells while less fluorescence was found in untreated cells.
Inhibition of autophagy could significantly enhance rhArg-induced cell growth inhibition
Given that rhArg could induce autophagy in leukemia cells, we explored whether rhArg-induced autophagy could affect the inhibitive efficacy of rhArg on leukemia cells. We employed 3-MA and CQ, two most extensively used pharmacological autophagy inhibitors, to inhibit rhArg-induced autophagy. 3-MA is a PI3K inhibitor and decreases autophagosomic LC3 expression to inhibit autophagy at an early stage [18]. CQ inhibits the fusion between autophagosomes and lysosomes to suppress autophagy at a later stage [19]. Confocal microscopy analyses showed that 3-MA inhibited autophagy fluorescence induced by rhArg in HL-60 and Jurkat leukemia cells. In contrast, CQ significantly enhanced immunofluorescence when compared with rhArg treatment alone (Fig. 4a, b). Compared with rhArg treatment alone, either 3-MA (1 or 2 mM) or CQ (10 or 20 μM) could remarkably strengthen rhArg-induced decrease of cell viability in HL-60 and Jurkat leukemia cells, respectively (Fig. 4c, d).

Inhibition of autophagy significantly enhanced rhArg-induced cell growth inhibition. a HL-60 cells were incubated with or without 1 IU/ml of rhArg in the presence or absence of the autophagy inhibitors 3-MA (1 mM) or CQ (10 μM) for 48 h. Cell samples were prepared for confocal microscopy analysis as described in “Materials and methods.” Untreated cells were used as negative control. b Jurkat cells were incubated with or without 1 IU/ml of rhArg in the presence or absence of the autophagy inhibitors 3-MA (1 mM) or CQ (10 μM) for 48 h. Cell samples were prepared for confocal microscopy analysis as described in “Materials and methods.” Untreated cells were used as negative control. c HL-60 cells were incubated with or without 1 IU/ml of rhArg in the presence or absence of the autophagy inhibitors 3-MA (1 or 2 mM) or CQ (10 or 20 μM) for 48 h. Cell growth inhibition was analyzed by MTT. The data are presented as the means ± SD of four samples. **p < 0.01 versus rhArg. A typical result from three independent experiments is presented. d Jurkat cells were incubated with or without 1 IU/ml of rhArg in the presence or absence of the autophagy inhibitors 3-MA (1 or 2 mM) or CQ (10 or 20 μM) for 48 h. Cell growth inhibition was analyzed by MTT. The data are presented as the means ± SD of four samples. **p < 0.01 versus rhArg. A typical result from three independent experiments is presented
Inhibition of autophagy could significantly enhance rhArg-induced cell apoptosis
To further understand the biological role of autophagy in rhArg-induced cell death and elucidate proapoptotic effects of rhArg after inhibiting autophagy, we examined the changes of rhArg-induced apoptosis in HL-60 and Jurkat leukemia cells. As shown in Fig. 5a, the proportion of Annexin V-positive cells was obviously increased in rhArg combined with 3-MA or CQ-treated cells. The results demonstrated that rhArg in combination with 3-MA or CQ induced a significant higher percentage of apoptotic cells when compared with rhArg treatment alone, while either 3-MA or CQ treatment alone rarely showed enhancement of apoptosis of leukemia cells (Fig. 5a, b). All these data demonstrated that autophagy acted as a cellular protective mechanism in rhArg-treated leukemia cells and suggested that targeting autophagy might encourage the therapeutic effects of rhArg on leukemia.

Inhibition of autophagy obviously enhanced rhArg-induced cell apoptosis. a HL-60 and Jurkat cells were incubated with or without 1 IU/ml of rhArg in the presence or absence of the autophagy inhibitors 3-MA (1 mM) or CQ (10 μM) for 48 h. Cells were stained with Annexin V/PI and analyzed by flow cytometry. The percentages of Annexin V-positive cells are shown in the bar charts (b). **p < 0.01 versus control or rhArg
Discussion
Leukemias are clonal malignancies of the hematopoietic system characterized by the accumulation of immature cell populations in the bone marrow or peripheral blood [20]. Although intensified multi-agent chemotherapy protocols have been developed for the treatment of leukemia, the high toxicity and therapeutic resistance still limited the uses of these drugs [21, 22]. So, it is urgent to investigate new agents or combinational therapies to solve the current condition of leukemia treatment.
The different metabolism between cancer cells and normal cells might become a treasure trove to discover new anti-tumoral strategies [23, 24]. Although arginine is a semi-essential amino acid to normal cells owing to its self-synthesis from citrulline, it is essential for most carcinoma cells which lack argininosuccinate synthetase or ornithine transcarbamylase to synthesize arginine [25, 26]. Thus, targeting arginine degradation might be a potential strategy for tumor treatment. Among arginine-depleting therapeutics, rhArg is being investigated in phase II clinical trials for HCC therapy and a variety of solid tumors as well as hematological malignancies have been reported sensitive to rhArg treatment [27]. Researchers have confirmed that rhArg could induce cytotoxicity in non-Hodgkin’s lymphoma cells, malignant melanoma cells, and triple-negative breast cancer cells [8, 11, 15]. Some therapeutic agents inhibit the growth of tumor cells via interfering with DNA replication progression, and cell cycle arrest in the sub-G1, G0, and G1 phases could lead to apoptosis [28, 29]. To investigate whether rhArg have therapeutic effects on leukemia cells, the growth inhibitory effects and changes of cell cycle distribution induced by rhArg were detected in HL-60 and Jurkat leukemia cells: rhArg could significantly inhibit leukemia cell proliferation and induce cell cycle arrest in the G0/G1 phase.
Autophagy is an evolutionarily conserved process through which cells recruit damaged proteins and dysfunctional organelles to autophagosomes and then fuse with lysosomes for their degradation under stress conditions such as hypoxia, nutrient stress, genomic instability, endoplasmic reticulum stress, and intracellular infections [30, 31]. Our previous study reported that rhArg could induce autophagy in lymphoma cells and autophagy played a crucial role in rhArg-induced lymphoma cell death [11]. In this study, the formation of autophagosomes and the conversion of LC3-I to LC3-II were explored to ensure autophagy induced by rhArg in leukemia cells. A growing body of literature has revealed that autophagy might promote either cell survival or cell death depending on different stimuli or the intracellular environment [32, 33]. To investigate whether autophagy could affect the inhibitory efficacy of rhArg on leukemia cells, 3-MA and CQ were used to inhibit autophagy induced by rhArg. 3-MA or CQ could significantly enhance the growth inhibitory and proapoptotic effects triggered by rhArg. Thus, inhibition of autophagy might be a potential strategy to enhance the anti-leukemia efficacy of rhArg-based therapy. As the autophagy inhibitor CQ has been approved by the FDA for clinical use, targeting arginine depletion and autophagy might encourage the therapy of leukemia in clinics [34].
In conclusion, our studies demonstrated that rhArg induced cytotoxicity in human leukemia cells in vitro: rhArg induced cell growth inhibition, cell cycle arrest in the G0/G1 phase, and apoptosis in HL-60 and Jurkat cells. Moreover, the combination of rhArg with autophagy inhibitors obviously promoted leukemia cell death, which confirmed the protective role of autophagy in rhArg-induced leukemia cell death and encouraged the application of autophagy agents for the anti-leukemia efficacy of rhArg in clinics.
References
Schmiegelow K, Hjalgrim H. Is the risk of acute lymphoblastic leukemia reduced in siblings to children with the disease? A novel hypothesis explored by international collaboration. Leukemia. 2006;20(7):1206–8. doi:10.1038/sj.leu.2404250.
Fan J, Dong X, Zhang W, Zeng X, Li Y, Sun Y, et al. Tyrosine kinase inhibitor Thiotanib targets Bcr-Abl and induces apoptosis and autophagy in human chronic myeloid leukemia cells. Appl Microbiol Biotechnol. 2014;98(23):9763–75. doi:10.1007/s00253-014-6003-1.
Kersey JH. Fifty years of studies of the biology and therapy of childhood leukemia. Blood. 1998;92(5):1838.
Christiansson L, Soderlund S, Mangsbo SM, Hjorth-Hansen H, Hoglund M, Markevarn B, et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol Cancer Ther. 2015;14(5):1181–91. doi:10.1158/1535-7163.MCT-14-0849.
Ma D, Fang Q, Wang P, Gao R, Wu W, Lu T, et al. Induction of heme oxygenase-1 by Na+−H+ exchanger 1 protein plays a crucial role in imatinib-resistant chronic myeloid leukemia cells. J Biol Chem. 2015;290(20):12558–71. doi:10.1074/jbc.M114.626960.
Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z, et al. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget. 2015;6(6):3861–73. doi:10.18632/oncotarget.2869.
Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell. 2014;56(2):205–18. doi:10.1016/j.molcel.2014.08.018.
Wang Z, Shi X, Li Y, Zeng X, Fan J, Sun Y, et al. Involvement of autophagy in recombinant human arginase-induced cell apoptosis and growth inhibition of malignant melanoma cells. Appl Microbiol Biotechnol. 2014;98(6):2485–94. doi:10.1007/s00253-013-5118-0.
Chow AK, Ng L, Sing Li H, Cheng CW, Lam CS, Yau TC, et al. Anti-tumor efficacy of a recombinant human arginase in human hepatocellular carcinoma. Curr Cancer Drug Targets. 2012;12(9):1233–43.
Lam TL, Wong GK, Chow HY, Chong HC, Chow TL, Kwok SY, et al. Recombinant human arginase inhibits the in vitro and in vivo proliferation of human melanoma by inducing cell cycle arrest and apoptosis. Pigment Cell Melanoma Res. 2011;24(2):366–76. doi:10.1111/j.1755-148X.2010.00798.x.
Zeng X, Li Y, Fan J, Zhao H, Xian Z, Sun Y, et al. Recombinant human arginase induced caspase-dependent apoptosis and autophagy in non-Hodgkin’s lymphoma cells. Cell Death Dis. 2013;4, e840. doi:10.1038/cddis.2013.359.
Qiu F, Chen YR, Liu X, Chu CY, Shen LJ, Xu J, et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci Signal. 2014;7(319):ra31. doi:10.1126/scisignal.2004761.
Zeng X, Zhao H, Li Y, Fan J, Sun Y, Wang S, et al. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy. 2015;11(2):355–72. doi:10.4161/15548627.2014.994368.
Li Y, Zhu H, Zeng X, Fan J, Qian X, Wang S, et al. Suppression of autophagy enhanced growth inhibition and apoptosis of interferon-beta in human glioma cells. Mol Neurobiol. 2013;47(3):1000–10. doi:10.1007/s12035-013-8403-0.
Wang Z, Shi X, Li Y, Fan J, Zeng X, Xian Z, et al. Blocking autophagy enhanced cytotoxicity induced by recombinant human arginase in triple-negative breast cancer cells. Cell Death Dis. 2014;5, e1563. doi:10.1038/cddis.2014.503.
Li Y, Zeng X, Wang S, Sun Y, Wang Z, Fan J, et al. Inhibition of autophagy protects against PAMAM dendrimers-induced hepatotoxicity. Nanotoxicology. 2015;9(3):344–55. doi:10.3109/17435390.2014.930533.
Wang S, Li Y, Fan J, Wang Z, Zeng X, Sun Y, et al. The role of autophagy in the neurotoxicity of cationic PAMAM dendrimers. Biomaterials. 2014;35(26):7588–97. doi:10.1016/j.biomaterials.2014.05.029.
Young C, Sinadinos A, Lefebvre A, Chan P, Arkle S, Vaudry D, et al. A novel mechanism of autophagic cell death in dystrophic muscle regulated by P2RX7 receptor large-pore formation and HSP90. Autophagy. 2015;11(1):113–30. doi:10.4161/15548627.2014.994402.
Hori YS, Hosoda R, Akiyama Y, Sebori R, Wanibuchi M, Mikami T, et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J Neurooncol. 2015;122(1):11–20. doi:10.1007/s11060-014-1686-9.
Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. 2000;19(21):2496–504. doi:10.1038/sj.onc.1203486.
Bassan R, Masciulli A, Intermesoli T, Audisio E, Rossi G, Pogliani EM, et al. Randomized trial of radiation-free central nervous system prophylaxis comparing intrathecal triple therapy with liposomal cytarabine in acute lymphoblastic leukemia. Haematologica. 2015;100(6):786–93. doi:10.3324/haematol.2014.123273.
Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet. 2012;3:249. doi:10.3389/fgene.2012.00249.
Tanaka K, Sasayama T, Irino Y, Takata K, Nagashima H, Satoh N, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. 2015;125(4):1591–602. doi:10.1172/JCI78239.
Tiwana GS, Prevo R, Buffa FM, Yu S, Ebner DV, Howarth A, et al. Identification of vitamin B1 metabolism as a tumor-specific radiosensitizing pathway using a high-throughput colony formation screen. Oncotarget. 2015;6(8):5978–89. doi:10.18632/oncotarget.3468.
Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, et al. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer. 2004;100(4):826–33. doi:10.1002/cncr.20057.
Mussai F, Egan S, Higginbotham-Jones J, Perry T, Beggs A, Odintsova E, et al. Arginine dependence of acute myeloid leukaemia blast proliferation: a novel therapeutic target. Blood. 2015;125(15):2386–96. doi:10.1182/blood-2014-09-600643.
Khoury O, Ghazale N, Stone E, El-Sibai M, Frankel AE, Abi-Habib RJ. Human recombinant arginase I (Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human glioblastoma cells. J Neurooncol. 2015;122(1):75–85. doi:10.1007/s11060-014-1698-5.
Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–8. doi:10.1038/35077213.
Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9(5), e98207. doi:10.1371/journal.pone.0098207.
Moreau P, Moreau K, Segarra A, Tourbiez D, Travers MA, Rubinsztein DC, et al. Autophagy plays an important role in protecting Pacific oysters from OsHV-1 and Vibrio aestuarianus infections. Autophagy. 2015;11(3):516–26. doi:10.1080/15548627.2015.1017188.
Wan G, Xie W, Liu Z, Xu W, Lao Y, Huang N, et al. Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy. 2014;10(1):70–9. doi:10.4161/auto.26534.
Chen P, Hu T, Liang Y, Jiang Y, Pan Y, Li C, et al. Synergistic inhibition of autophagy and neddylation pathways as a novel therapeutic approach for targeting liver cancer. Oncotarget. 2015;6(11):9002–17. doi:10.18632/oncotarget.3282.
Vogel RI, Coughlin K, Scotti A, Iizuka Y, Anchoori R, Roden RB, et al. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget. 2015;6(6):4159–70. doi:10.18632/oncotarget.2904.
Lotze MT, Buchser WJ, Liang X. Blocking the interleukin 2 (IL2)-induced systemic autophagic syndrome promotes profound antitumor effects and limits toxicity. Autophagy. 2012;8(8):1264–6. doi:10.4161/auto.20752.
Acknowledgments
This work was supported by grants from the National Key Basic Research Program of China (2013CB932502, 2015CB931800), the National Natural Science Foundation of China (81573332), and Shanghai Science and Technology Funds (14431900200).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
None
Additional information
Yubin Li and Xian Zeng contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
rhArg induced formation of autophagosomes in leukemia cells. HL-60 and Jurkat cells were treated with or without 1 IU/ml of rhArg for 24 h. Cell samples were prepared for transmission electron microscopy analysis as described in “Materials and methods.” Autophagosomes were counted, and the data were presented as the means ± SD of four samples. **p < 0.01 versus Ctrl; ***p < 0.001 versus Ctrl (JPEG 19 kb)
Rights and permissions
About this article

Cite this article
Li, Y., Zeng, X., Wang, S. et al. Blocking autophagy enhanced leukemia cell death induced by recombinant human arginase. Tumor Biol. 37, 6627–6635 (2016). https://doi.org/10.1007/s13277-015-4253-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4253-x