
Abstract
Violacein (VIO; 3-[1,2-dihydro-5-(5-hydroxy-1H-indol-3-yl)-2-oxo-3H-pyrrol-3-ylidene]-1,3-dihydro-2H-indol-2-one), an indole-derived purple-colored pigment, produced by a limited number of Gram-negative bacteria species, including Chromobacterium violaceum and Janthinobacterium lividum, has been demonstrated to have anti-cancer activity, as it interferes with survival transduction signaling pathways in different cancer models. Head and neck carcinoma (HNC) represents the sixth most common and one of the most fatal cancers worldwide. We determined whether VIO was able to inhibit head and neck cancer cell growth both in vitro and in vivo. We provide evidence that VIO treatment of human and mouse head and neck cancer cell lines inhibits cell growth and induces autophagy and apoptosis. In fact, VIO treatment increased PARP-1 cleavage, the Bax/Bcl-2 ratio, the inhibition of ERK1 and ERK2 phosphorylation, and the expression of light chain 3-II (LC3-II). Moreover, VIO was able to induce p53 degradation, cytoplasmic nuclear factor kappa B (NF-κB) accumulation, and reactive oxygen species (ROS) production. VIO induced a significant increase in ROS production. VIO administration was safe in BALB/c mice and reduced the growth of transplanted salivary gland cancer cells (SALTO) in vivo and prolonged median survival. Taken together, our results indicate that the treatment of head and neck cancer cells with VIO can be useful in inhibiting in vivo and in vitro cancer cell growth. VIO may represent a suitable tool for the local treatment of HNC in combination with standard therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Violacein (VIO; 3-[1,2-dihydro-5-(5-hydroxy-1H-indol-3-yl)-2-oxo-3H-pyrrol-3-ylidene]-1,3-dihydro-2H-indol-2-one) is an indole-derived purple-colored pigment, produced by a limited number of Gram-negative bacteria species, including Chromobacterium violaceum and Janthinobacterium lividum. The role of VIO in bacterial life and metabolism is not fully understood. Different studies have demonstrated a potential role for VIO in environmental competition with other microorganisms, as it has anti-protozoal, anti-bacterial, and anti-Candida activities [1–8]. These properties may play a key role in the survival and diffusion of VIO-producing bacteria in ecological competition with other microorganisms for nutrients. Thus, VIO production and biofilm development may represent a response to environmental stresses and a key factor in survival mechanisms of J. lividum. As a consequence, VIO and biofilm production, both under quorum-sensing regulation, could be considered to be beneficial factors of J. lividum resistance to different types of environmental stress [9].
Several studies have demonstrated that VIO has anti-tumor, anti-viral, anti-ulcerogenic, immunomodulatory, analgesic, and anti-pyretic activities [10]. Indeed, it has been shown that VIO induces apoptosis in HL60 leukemic cells through the activation of caspase-8, the transcription of nuclear factor kappa B (NF-κB) target genes and the activation of p38 mitogen-activated protein (MAP) kinase and the tumor necrosis factor alpha (TNF-α) signal transduction pathway. In addition, VIO was shown to be non-cytotoxic in normal healthy cells [7]. The anti-tumor potential of VIO is not restricted to acute myeloblastic leukemia. Recent reports have demonstrated its beneficial effects against BCR-ABL-positive chronic myeloid leukemia cells (K562 cell line), including their multidrug-resistant variant. The molecular mechanisms of VIO action involve the activation of specific signaling transduction pathways, dependent on the cell type. This observation was corroborated by studies performed in prostate, lung, colon cancer cell lines and in Ehrlich’s ascites tumors [11–13]. In addition, it has been demonstrated that VIO was highly cytotoxic for colorectal cancer (CRC) cells [12]. VIO inhibited Akt phosphorylation with the subsequent reactivation of the apoptosis pathway and downregulation of NF-κB, MDM2, p21–p27, GSK3α/β and Bad signaling in CRC cells [13]. Recently, it has been reported that VIO inhibited the proteolytic activity of MMPs and downregulated CXCL12–CXCR4 interactions, thus inhibiting cell migration and invasion in breast cancer cell lines [14]. VIO was shown to increase the production of reactive oxygen species (ROS), followed by the activation of caspase-3, the release of cytochrome c, the release of calcium from the cytosol and the induction of the collapse of the mitochondrial membrane, thus leading to tumor cell apoptosis [11].
There is limited evidence for VIO having an effect on solid tumors, both in vitro and in vivo. Head and neck carcinoma (HNC) represents the sixth most common cancer worldwide, and it mainly involves the larynx, pharynx, oral cavity, and tongue [15, 16].
The treatments for HNC have been associated with considerable complications and potential treatment-related death. Thus, although there have been significant advances in both surgical and non-surgical therapy, the overall survival and quality of life of patients with HNC have not been significantly improved over the past decade, especially for patients in an advanced stage. Indeed, the current strategies to cure these tumors have limited efficacy and documented toxicity and HNC remains one of the most fatal cancers worldwide [17]. The discovery and development of innovative drugs might supplement the pharmaceutical armamentarium presently used for HNC management.
Cancer chemoprevention makes use of natural or synthetic compounds to prevent, arrest, or reverse the process of carcinogenesis in its earliest stages [18]. Moreover, compounds derived from bacteria are now being tested for their anti-cancer activity [19, 20].
This study was aimed at determining VIO anti-tumor activities in HNC both in vitro and in vivo. In this study, we provide evidence that VIO inhibits proliferation and induces autophagy and apoptosis of HNC cell lines in vitro and delays the tumor cell growth of a salivary gland carcinoma cell line in vivo. Elucidation of the molecular mechanism mediating this activity provides further relevant information for understanding its anti-tumoral effects and for considering its possible clinical applications.
Materials and methods
Reagents
DMSO, sulforhodamine B (SRB), and Hoechst 33342 were purchased from Sigma-Aldrich (Milan, Italy). Rabbit polyclonal anti-Bax and mouse monoclonal anti-Bcl-2 antibodies were obtained from BD Pharmingen (BD Biosciences). Antibodies against ERK1/2 (C-14), phospho-ERK (E-4), NF-κB, PARP-1, and p53 (DO-1) were obtained from Santa Cruz Biotechnology (CA, USA). Antibody against light chain 3 (LC3) was obtained from Novus Biologicals (Littleton, CO, USA). Rabbit polyclonal anti-actin was from Sigma-Aldrich. The anti-caspase-9 and anti-caspase-8 antibodies were purchased from Cell Signaling Technology (MA, USA). Goat anti-mouse IgG Alexa Fluor-488-conjugated antibody was purchased from Life Technologies™ Molecular Probes (Oregon, USA), and the goat anti-mouse or anti-rabbit IgG peroxidase-conjugated secondary antibodies were from Sigma-Aldrich.
Production of VIO
For VIO production and extraction, the bacterial strain J. lividum DSM1522T was used. J. lividum was maintained frozen in a brain–heart infusion broth (BHI; Difco Laboratories, Livonia, MI, USA) containing glycerol (25 %) at −80 °C until use. J. lividum was cultured and checked for purity on trypticase soy agar (TSA; Oxoid Ltd., Basingstoke, UK) at 25 °C for 48 h. To obtain VIO production, J. lividum DSM1522T was cultured in a modified Luria-Bertani (LB) broth medium supplemented with 1 % glycerol as carbon source (LBY) [9]. Briefly, after checking for purity on a TSA agar plate, a single colony was taken as a pre-inoculum in LBY maintained at 25 °C until reaching the logarithmic phase of growth.
Pre-inoculum (1:100 v/v) was used to inoculate 5 l of LBY, distributed in culture flasks (50 cc each; Corning® Erlenmeyer cell culture flasks with venting position), and incubated in static conditions at 25 °C until reaching the late stationary phase. After 48 h of incubation, the growth medium was removed by centrifugation, and the purple-colored pellet of cells was re-suspended in absolute methanol (three fourth of the initial volume), gently mixed by hand for 15 min and then centrifuged at 13,000 rpm for 30 min. After centrifugation, the pellet containing cells and cell debris was discarded and the supernatant containing the eluted pigment VIO was recovered for further purification. To obtain a final product with fewer impurities, we only recovered pigment from cells.
Purification of VIO
All reagents and solvents were handled according to the material safety data sheet of the supplier and were used as purchased. Macherey-Nagel silica gel pre-coated aluminum cards with a fluorescent indicator visualizable at 254 nm were used for thin-layer chromatography. Developed plates were visualized with a Spectroline ENF-260C/FE UV apparatus. Evaporation of the solvents was carried out on a Büchi Rotavapor R-210 equipped with a Büchi V-855 vacuum controller and Büchi V-700 (~5 mbar) and V-710 (~2 mbar) vacuum pumps. Flash chromatography was carried out with an Interchim Spot II Flash, using Merck SuperVarioFlash D26 cartridges packed with Merck Geduran 60 (0.040–0.063 mm) silica gel. Melting points (MPs) were determined using a Stuart Scientific SMP1 apparatus and are uncorrected. Infrared (IR) spectra were run on PerkinElmer Spectrum One FT-ATR spectrophotometer. Proton (1H, 400.13 MHz) and carbon (13C, 100.6 MHz) nuclear magnetic resonance spectra were recorded by a Bruker Avance 400. 1H and 13C nuclear magnetic resonance (NMR) data were acquired by Bruker TopSpin 2.1 software and processed by Mestrelab Research S.L. MestreReNov. 6.2.1–769 software. Chemical shifts are expressed in delta units (ppm) with tetramethylsilane. The purity of tested compounds was found to be >95 % by high-pressure liquid chromatography (HPLC) analysis. The HPLC system that was used (Thermo Fisher Scientific Inc., Dionex UltiMate 3000) comprised a SR-3000 solvent rack, a LPG-3400SD quaternary analytical pump, a TCC-3000SD column compartment, a DAD-3000 diode array detector, and an analytical manual injection valve with a 20-μl loop. Samples were dissolved in acetonitrile at 1 mg/ml. HPLC analysis was performed with a Thermo Fisher Scientific Inc., Acclaim 120 C18 reversed-phase column (5 μm, 4.6 mm × 250 mm) at 25 ± 1 °C with an isocratic gradient (acetonitrile:water = 90:10), flow rate of 1.0 ml/min, and signal detector at 254 and 365 nm. Chromatographic data were acquired and processed by Thermo Fisher Scientific Inc., Chromeleon 6.80 software.
The methanol extract (≈550 ml) was evaporated in vacuo, producing a black residue (0.87 g). The crude product was purified by flash chromatography (chloroform:ethanol as eluent) to produce VIO as a purple solid (7.3 mg), MP > 300 °C (from methanol), and literature MP > 350 °C [21]. 1H NMR, 13C NMR, and IR spectra were identical to those reported in the literature [21].
Cell lines and treatments
Cell lines derived from carcinoma of the tongue (CAL-27 and SCC-15) or pharynx (FaDu) were maintained in RPMI containing 10 % fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. MCF-10A (a non-tumorigenic epithelial cell line) and HaCaT (a human immortalized keratinocyte cell line) cells were maintained in Dulbecco’s modified eagle medium (DMEM) containing 10 % fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. For treatments, cells were incubated for the indicated times in the presence of VIO (dose range 1–15 μM) or vehicle control (DMSO ≤ 0.1 %). Neu-overexpressing salivary gland cancer cells (H-2d) (SALTO) were kindly provided by Prof. F. Cavallo (University of Torino) and Prof. P.L. Lollini (University of Bologna) and maintained in DMEM containing 20 % fetal bovine serum (FBS). SALTO cells were established from salivary carcinoma arising in BALB-neuT transgenic male mice hemizygous for p53172R-H transgene driven by the whey acidic protein promoter [22].
SRB assay
Cells were seeded at 4 × 103/well in 96-well plates and incubated at 37 °C to allow cell attachment. After 24 h, the medium was changed and the cells were treated with VIO or with DMSO and incubated for 24, 48, or 72 h. Cells were then fixed with cold trichloroacetic acid (final concentration 10 %) for 1 h at 4 °C. The assay was then performed as previously described [23]. The percentage survival of the cultures treated with the compounds or DMSO was calculated by normalization of their optical density (OD) values to those of untreated control cultures [23]. The experiments were performed in triplicate and repeated three times.
Fluorescence-activated cell sorting analysis
Asynchronized log phase growing cells (60 % confluent, approximately 2.5 × 105/well in 6-well plates) were treated with VIO or with DMSO in complete culture medium. After 48 h, adherent as well as suspended cells were harvested, centrifuged at 1500 rpm for 10 min and washed twice with cold phosphate-buffered saline (PBS). The assay was then performed as previously described [24]. Cells were analyzed by flow cytometry using a fluorescence-activated cell sorting (FACS) Calibur cytometer running CellQuest software.
Western blotting
Briefly, 1 × 106 cells were seeded in 100-mm tissue culture dishes 24 h prior to the addition of 5 or 7.5 μM of VIO or DMSO. After 24 h of treatment, the cells were harvested, washed twice with cold PBS, and lysed in RIPA buffer, as previously described [24]. For immunoblotting analysis, 50–80 μg of cell lysates were resolved by SDS-PAGE and then transferred to nitrocellulose membranes. Equal loading and transfer of proteins were verified by Ponceau red staining of the filters and actin immunostaining. The assay was then performed as previously described [24].
Fluorescent measurement of ROS
Dichlorofluorescin diacetate (DCF-DA) was used to detect ROS production in cells. Briefly, 2.5 × 105 cells were seeded into 6-well plates and incubated at 37 °C to allow cell attachment before treatment. After two washings with PBS, cells were incubated with 10 μM 2′,7′-dichlorofluoresce in diacetate (Sigma-Aldrich, Milan, Italy) in PBS at 37 °C and 5 % CO2 in the dark for 30 min [25]. After two washings, cells were treated with VIO in serum-free medium and incubated at 37 °C and 5 % CO2 in the dark for different times (15 min–4 h). Then, adherent cells and suspended cells were harvested, centrifuged at 1250 rpm for 10 min, and seeded in 96-well plates (100 μl per well). Fluorescence intensity was measured after 15 and 30 min and 1 and 4 h using a spectrophotometric plate reader at an excitation wavelength of 495 nm and an emission wavelength of 535 nm. Because the highest level of fluorescence was detected at 30 min and then decreased back to the level of the control after 1 h of stimulation (data not shown), this experimental time was chosen for subsequent experiments.
Immunofluorescence
Cells were seeded (2.5 × 104/well) on eight-chamber Permanox slides (Lab-Tek, IL) in 300 μl of culture medium, grown for 24 h, and treated with VIO or with DMSO in complete culture medium. After 24 h, the culture medium was removed, and after two washes with PBS, the cells were fixed with cold methanol at −20 °C for 10 min [26]. The cells were then incubated at room temperature with primary antibodies for 1 h and, after two further washes, with goat anti-mouse IgG Alexa Fluor-488-conjugated secondary antibody for 45 min. Nuclei were counterstained with Hoechst 33342. The slides were then mounted with glycerol, observed with an Olympus BX51 fluorescence microscope, and analyzed with IAS software.
Treatment of BALB/c mice with VIO
BALB/c mice were subcutaneously injected in the right flank with a 0.2 ml suspension containing 1 × 106 SALTO cells in PBS. Groups of BALB/c mice (five or six mice per group) were treated weekly, starting 3 weeks after SALTO transplantation, with an intratumoral injection of 0.75 mg/kg of VIO dissolved in DMSO and diluted in PBS. The control group was intratumorally injected with DMSO dissolved in PBS.
Mice were sacrificed at the first signs of distress. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) and carried out according to Italian rules (D.L.vo 116/92 and C.E. 609/86). A veterinary surgeon was present during the experiments. Animal care, before and after the experiments, was carried out only by trained personnel.
Analysis of anti-tumor activity in vivo
Tumor growth was monitored weekly until tumor-bearing mice were sacrificed when the tumor exceeded 20 mm in width. Tumors were measured by a caliper in two dimensions, and the volumes were calculated using the following formula: width2 × length/2 [27].
Statistical analysis
Data distribution of cell survival and FACS analyses were preliminarily verified by the Kolmogorov-Smirnov test, and data sets were analyzed with a one-way analysis of variance (ANOVA) followed by a Newman-Keuls test. Survival curves and tumor volumes were estimated using the Kaplan-Meier method and compared with a log-rank test with calculation of the SD according to the method of Greenwood. Differences were regarded as significant when the p value was ≤0.05 [28].
Results
Production and purification of VIO
To produce VIO, J. lividum DSM1522T was cultured in LBY. Pre-inoculum (1:100 v/v) was used to inoculate 5 l of LBY, and it was incubated in static conditions at 25 °C until reaching the late stationary phase. After 48 h of incubation, the growth medium was removed by centrifugation and the purple-colored pellet of cells was re-suspended in absolute methanol. From 5 l of LBY culture of J. lividum DSM1522T, approximately 3 l of raw VIO was recovered and extracted using absolute methanol as the solvent. The extracted VIO, quantified by a spectrophotometer (OD585 = 5.81), was used for subsequent purification. Part of the methanol extract was evaporated and purified by flash chromatography to obtain pure VIO.
Inhibition of human and mouse HNC cells survival by VIO
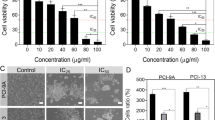
To evaluate the effect of VIO on the survival of tongue (CAL-27 and SCC-15) and pharynx (FaDu) carcinoma cell lines, increasing concentrations (1, 2.5, 5, 10, and 15 μM) of VIO or vehicle control (DMSO) cells were incubated for 24, 48, and 72 h. The effect of VIO on cell growth of mouse SALTO cells was analyzed as well. The effect on cell growth was determined with the SRB assay. As shown in Fig. 1, VIO significantly inhibited the growth of all cancer cell lines in a dose- and time-dependent manner. Indeed, better inhibition of cell proliferation was obtained for all cell lines treated with VIO compared to those treated with DMSO (p < 0.001) at concentrations of 10–15 μM and after 72 h. However, FaDu and SALTO cells were sensitive to VIO treatment even at the lower concentrations at 24 h (p < 0.01 for FaDu cells treated with 1 and 2.5 μM VIO and p < 0.05 and p < 0.01 for SALTO cells treated with 1 and 2.5 μM VIO, respectively; Fig. 1). The concentration of VIO required to inhibit cell survival by 50 % (IC50) ranged between 2.32 and 7.06 μM after 48 h of treatment (Table 1).

Effect of VIO on head and neck carcinoma cell line survival. Survival of human pharynx (FaDu), tongue (CAL-27 and SCC-15), and mouse salivary gland (SALTO) cancer cell lines and human non-tumorigenic epithelial (MCF-10A) and immortalized keratinocyte (HaCaT) cell lines were assessed with a SRB assay after 24, 48, or 72 h of treatment with DMSO or VIO. The results are reported as the mean ± SD values from three experiments performed in triplicate (§p < 0.05, *p < 0.01, #p < 0.001 vs DMSO treatment). **Significant difference in cell survival between HNC and MCF-10A cells, FaDu versus MCF-10A, p < 0.001(15, 10, 5, and 2.5 μM at 24 h; 15, 10, and 5 μM at 48 h; and 15–10 μM at 72 h), p < 0.01 (1 μM at 24 h), and p < 0.05 (5 μM at 72 h); CAL-27 versus MCF-10A, p < 0.001 (15, 10, and 5 μM at 24, 48, and 72 h) and p < 0.05 (2.5 μM at 24 h); and SCC-15 versus MCF-10A, p < 0.001 (15–10 μM at 24 h and 15, 10, and 5 μM at 48 and 72 h) and p < 0.01 (5 μM at 24 h). **Significant difference in cell survival between HNC and HaCaT cells, FaDu versus HaCaT, p < 0.001 (all doses at 24 h; 15, 10, and 5 μM at 48 h; and 15–10 μM at 72 h), p < 0.01 (5 μM at 72 h), and p < 0.05 (2.5–1 μM at 48 h); CAL-27 versus HaCaT, p < 0.001 (15, 10, and 5 μM at 24, 48, and 72 h) and p < 0.01 (2.5 μM at 24 h); and SCC-15 versus HaCaT, p < 0.001 (15–10 μM at 24 h; 15, 10, and 5 μM at 48 and 72 h) and p < 0.01 (5 μM at 24 h)
In addition, the survival of a human non-tumorigenic epithelial cell line (MCF-10A), and of a human immortalized keratinocyte cell line (HaCaT), upon VIO treatment was evaluated. It is of note that VIO had no effects on survival of MCF-10A and HaCaT cells at the concentrations of 1–2.5 to 5–10 μM after 24, 48, or 72 h. However, VIO significantly inhibited cells survival after 48 and 72 h at the higher dose (15 μM) (p < 0.05 and p < 0.001, respectively, for MCF-10A and p < 0.01 and p < 0.001, respectively, for HaCaT), thus suggesting an in vitro toxic effect of the compound when used at this concentration. However, significant differences in cell survival between HNC and MCF-10A and HaCaT cells were also observed at the 15 μM concentration. Differences of cells survival between HNC and MCF-10A and HaCaT cells are reported in Fig. 1.
VIO induces apoptosis in HNC cell lines
The effect of VIO on the cell cycle distribution in HNC and MCF-10A cell lines was determined by flow cytometric analysis. Cells were incubated for 48 h with VIO (10 and 5 μM) or with DMSO as a control (Fig. 2). The mean results of three independent experiments are reported in Table 2.

Effect of VIO on cell cycle distribution. FACS analysis of DNA content was performed on asynchronized log phase growing HNC and MCF-10A cells treated for 48 h with DMSO or VIO at 5 and 10 μM. A representative experiment is shown in the figure
As shown in Table 2, treatment with VIO induced a dose-dependent increase in the percentage of cells in sub-G1 phase (p < 0.001 for FaDu, CAL-27, and SALTO and p < 0.01 for SCC-15 cells treated with 10 μM VIO) and a decrease in the percentage of cells in the G0/G1 phase (p < 0.001) in all cell lines. In addition, a decrease in the percentage of cells in the G2/M phase was observed in SCC-15, FaDu, and SALTO cells (p < 0.05 for SCC-15 and p < 0.01 for FaDu and SALTO cells) at the highest concentration. A simultaneous decrease in the percentage of cells in S-phase was observed in FaDu and SALTO cells (p < 0.05 for FaDu cells treated with 10 μM VIO and p < 0.001 and p < 0.01 for SALTO cells treated with 10 and 5 μM VIO, respectively), while CAL-27 showed an increase in the percentage of cells in this phase at 5 μM (p < 0.001). No effects of VIO on the cell cycle distribution were observed in MCF-10A cells at all doses tested (Fig. 2).
To demonstrate that the effect of VIO on the increase of cells in sub-G1 was due to the induction of apoptosis, we analyzed different signal transduction pathways involved in the activation of the apoptotic process.
HNC cell lines were treated with VIO (5–7.5 μM) for 24 h, and the expressions of Bax, Bcl-2, procaspase-9 and procaspase-8, active caspase-8, PARP-1, and p53 were analyzed by Western blotting. As shown in Fig. 3A, treatment with 5 μM VIO induced an increase of the expression of Bax (pro-apoptotic) in all lines examined and a decrease of Bcl-2 (anti-apoptotic) in CAL-27, SCC-15, and SALTO cells compared to DMSO-treated cells (p = 0.019, p = 0.048, and p = 0.045, respectively). Similar results were obtained using 7.5 μM of VIO (p = 0.042) in FaDu cells. The increased expression of Bax and the decreased expression of Bcl-2 determined by VIO treatment shifted the cell balance to programmed cell death.

Effect of VIO on cell apoptosis and autophagy. a Assessment of Bax, Bcl-2, procaspase-9, procaspase-8, caspase-8, cleavage of PARP-1 and LC3 levels by Western blotting in FaDu, CAL-27, SCC-15, and SALTO cells treated for 24 h with VIO at 5–7.5 μM or with DMSO as vehicle. Actin was used as an internal control. The intensities of the bands obtained in two independent experiments were quantified using ImageJ software after blot scanning, and the densitometric ratios between the Bax and Bcl-2, between procaspase-9 and actin, and between cleaved and full length PARP-1 are reported. Expression and densitometric quantization of LC3-II in HNC cells after treatment with VIO using Western blotting (p < 0.01). b Assessment of p53 levels by Western blotting in FaDu, CAL-27, and SCC-15 cells treated for 24 h with VIO at 5–7.5 μM or with DMSO as vehicle. Sub-cellular localization of p53 was analyzed by indirect immunofluorescence. After treatment, cells were fixed and incubated with anti-p53 antibody. After two washes with PBS, cells were incubated with the secondary Alexa Fluor-488-conjugated goat anti-mouse IgG antibody. Nuclei were stained with Hoechst 33342. Original magnification 400x
To determine which apoptotic pathway was activated after VIO treatment, the expressions of procaspase-9 and procaspase-8 and caspase-8 were analyzed with Western blotting in FaDu, CAL-27, SCC-15, and SALTO cells. VIO reduced the level of procaspase-9 compared to DMSO (p < 0.001), thus indicating its activation [29] (Fig. 3a). In addition, procaspase-8 was cleaved after VIO treatment.
Treatment with VIO at a concentration of 5 μM (CAL-27, SCC-15, and SALTO) or 7.5 μM (FaDu) was able to induce the proteolytic cleavage of PARP-1 in CAL-27 (p = 0.015), SCC-15 (p = 0.006), FaDu (p = 0.046), and SALTO (p = 0.044) cells, compared to DMSO-treated cells (Fig. 3a).
However, VIO-treated CAL-27 and SCC-15 showed a reduced expression of p53 compared to DMSO-treated cells (p < 0.05 and p < 0.01, respectively). All carcinoma cell lines showed the appearance of a lower molecular weight product, which suggests that treatment with VIO is able to induce p53 degradation (Fig. 3b). Analysis by fluorescence microscopy confirmed a decrease in p53 expression after VIO treatment compared to DMSO-treated cells. There was no change in the nuclear localization after VIO treatment (Fig. 3b).
VIO induces autophagy in HNC cells
Autophagy is considered a programmed cell death type II. The formation of an “autophagosome” (vacuole membrane with a double layer) is the marker of autophagy. The activation of the autophagic process can be detected by the conversion of LC3-I (a membrane protein autophagosome) in LC3-II, because the amount of LC3-II clearly correlated with the number of autophagosomes [30]. LC3-I (16 kDa) is a cytosolic protein, while LC3-II (14 kDa), which is the conjugated form with phosphatidylethanolamine (PE), is located in the membrane autophagosome. To evaluate the effect of VIO on the induction of autophagy, CAL-27, SCC-15, FaDu, and SALTO cells were treated with VIO or DMSO at a concentration of 5–7.5 μM for 24 h. As shown in Fig. 3a, LC3-I and LC3-II were constitutively expressed in DMSO-treated cells. After treatment with VIO, the expression of LC3-II was significantly enhanced compared to control cells (p < 0.01). Therefore, our results indicated that VIO treatment induced autophagy.
VIO inhibits mitogen-activated protein kinase signal and the NF-κB transduction pathway in HNC cells
The mitogen-activated protein kinase (MAPK) pathway is a signaling transduction pathway involved in cell growth and is often aberrantly activated in tumors. To evaluate whether VIO was able to inhibit MAPK activation, CAL-27, SCC-15, FaDu, and SALTO cells were treated with VIO or DMSO for 24 h and analyzed for the expression and activation of ERK1 and ERK2. As shown in Fig. 4a, treatment with VIO inhibited the level of phosphorylation of ERK1 in FaDu (p = 0.048), SCC-15 (p = 0.014), and SALTO (p = 0.023) cells, compared to DMSO-treated cells. VIO treatment inhibited phosphorylation of ERK2 in FaDu (p = 0.04), CAL-27 (p = 0.045), SCC-15 (p = 0.034), and SALTO (p = 0.006) cells.

Effect of VIO on pro-survival signaling proteins. a ERK1/ERK2 phosphorylation status in treated HNC cell lines. Western blotting was performed on VIO-treated (5–7.5 μM) or DMSO-treated cells for 24 h. The levels of phosphorylated ERK1/ERK2 were compared with those of the total ERK proteins, and the ratios are reported. b Expression and densitometric quantization of NF-κB in HNC cells after treatment with VIO by Western blotting. Inhibition of nuclear translocation of NF-κB after treatment with VIO in HNC cells. After treatment, cells were fixed and incubated with the anti-NF-κB antibody. Alexa Fluor-488-conjugated goat anti-mouse IgG antibody was used as secondary anti-body. Nuclei were stained with Hoechst 33342. Original magnification 400x
The transcription factor NF-κB promotes the activation of genes involved in inflammation, malignant transformation, tumor invasion, and angiogenesis. Because the activation of this transcription factor is associated with the process of carcinogenesis, its inhibition may be able to counteract neoplastic growth. CAL-27, SCC-15, and FaDu cells were treated with VIO or DMSO for 24 h and were then analyzed for the expression of NF-κB. A representative experiment is shown in Fig. 4. Treatment with VIO decreased the expression of NF-κB only in SCC-15 cell (p < 0.01; Fig. 4b). NF-κB functions as a transcription factor, moving into its active form in the nucleus. To determine if treatment with VIO was able to affect translocation of NF-κB to the nucleus, the localization of NF-κB was determined by immunofluorescence. NF-κB was found to be mainly localized in the nucleus in DMSO-treated FaDu, CAL-27, and SCC-15 cells. In contrast, treatment with VIO induced an accumulation of NF-κB in the cytoplasm, thus indicating an inhibitory effect of VIO on NF-κB nuclear translocation (Fig. 4b).
VIO induces ROS production in human HNC cells
To determine the effect of the compound at different concentrations (5, 10, and 20 μM) on intracellular ROS production in human HNC, a DCF-DA assay was performed. The effects of the compound were compared to those of DMSO, and the results were expressed as the fluorescence intensity (Table 3). VIO induced a significant increase in ROS production at a concentration of 20 μM in CAL-27 cell (p < 0.05) and at concentrations of 10 and 20 μM in FaDu cell (p < 0.05). Moreover, VIO induced a significant dose-dependent ROS production at all concentrations in SCC-15 cell (p < 0.05, p < 0.05, and p < 0.001, respectively; Table 3).
Delay of tumor growth in vivo by treatment with VIO
To evaluate whether the administration of VIO was able to inhibit the growth of transplanted SALTO cell, groups of BALB/c mice were treated by intratumoral injection with VIO starting 3 weeks after a tumor cell transplant. DMSO was used as a negative control. VIO reduced the mean tumor volume compared to control mice 1 week after the first VIO administration (157 vs 614 mm3; p < 0.0001) and maintained this decrease until 3 weeks following the beginning of treatment (418 vs 3078 mm3, respectively; p < 0.0001). At this stage (6 weeks post-transplant), DMSO-treated mice were sacrificed for exceeding tumor volume, while all VIO-treated mice remained alive at this time. Two VIO-treated mice were sacrificed 9 weeks after treatment, while four of these were still alive. All VIO-treated mice were sacrificed between weeks 12 and 13 after treatment. One VIO-treated mouse was still alive after 30 weeks with stable disease (320 mm3) (Fig. 5). VIO prolonged the median survival time compared to DMSO (12.5 vs 3 weeks) (p = 0.0005). Overall, the risk of developing tumors in DMSO-treated mice was 38.79 times greater than in the VIO-treated mice. Thus, our results suggest that VIO is able to significantly delay tumor growth and prolong median survival in comparison to DMSO (Table 4).

Delay of in vivo tumor growth by treatment with VIO. Groups of BALB/c mice were treated with VIO 3 weeks after SALTO tumor cell implantation. Differences in the tumor volumes and the mean survival time among the treated mice are reported
Discussion
Cancer chemoprevention employs natural or synthetic compounds to prevent, arrest, or reverse carcinogenesis [18]. The use of bacteria for the treatment of certain forms of cancer has been utilized since the early part of the last century [31]. In recent decades, compounds derived from bacteria have been tested for their anti-cancer activity [19, 20]. This involves the use of attenuated bacterial strains, spores, or bacterial strains engineered to express foreign genes. Novel approaches include the use of bacterial products such as proteins, enzymes, or secondary metabolites such as pigments, which can cause growth inhibition, cell cycle arrest, or the apoptosis of tumor cells [32]. Natural pigments can be purified from ores, insects, plants, and microbes. Among the others, bacteria have great potential to produce diverse pigments. Bacterial pigments can be easily produced and purified, and several strategies have been developed to improve the yield and quality of pigment production. Among the molecules produced by bacteria are carotenoids, melanins, flavins, phenazines, quinones, bacteriochlorophylls, monascins, violacein, and prodigiosin [33]. Among the others, prodigiosins and VIO have been demonstrated to have anti-cancer activity [32].
VIO (3-[1,2-dihydro-5-(5-hydroxy-1H-indol-3-yl)-2-oxo-3H-pyrrol-3-ylidene]-1,3-dihydro-2H-indol-2-one), an indole-derived purple-colored pigment, produced by a limited number of Gram-negative bacteria species, including C. violaceum and J. lividum [9], has been shown to display anti-cancer activity by interfering with survival signaling pathways in different cancer models [10–14].
HNC represents the sixth most common cancer worldwide and mainly involves the larynx, pharynx, oral cavity, and tongue [15, 16]. The current strategies to cure HNC have limited efficacy and documented toxicity, and HNC remains one of the most fatal cancers worldwide [17]. The prognosis of HNC is discouraging. Local tumor progression leads to morbidity and even death in the majority of patients. Chemotherapy regimens used in these patients show very significant side effects and a poor quality of life [34].
Intratumoral delivery of cancer therapeutics promises to be a good route of administration for such agents in easily accessible tumors. Several pre-clinical and phase I and phase II clinical trials are employing intratumoral delivery to deliver different therapeutics such as drugs, viral-based cancer vaccines, immune cell-based vaccines, cytokines, DNA, bacterial products, and natural compounds to the tumor site [28, 35–39]. An intratumoral route of administration is able to prevent the occurrence of systemic side effects and makes the therapeutic agents directly available at the tumor site, allowing for the highest concentration close to tumor cells.
In this study, we provided evidence that VIO inhibits HNC cell proliferation and induces apoptosis in HNC cell lines. Our results indicate that VIO is able to inhibit the growth of different cell lines in a dose-dependent manner. Moreover, treatment with VIO induced a dose-dependent increase in the percentage of cells in the sub-G1 phase, an increased expression of Bax, a decreased expression of Bcl-2 and PARP-1 cleavage. VIO treatment was able to activate both the intrinsic and extrinsic apoptotic pathways, as demonstrated by the cleavage of procaspase-8 and the decreased expression of procaspase-9. It has been previously demonstrated that VIO is able to induce the activation of both the intrinsic and extrinsic apoptotic pathways in colorectal cancer and leukemia cells [13, 40]. VIO did not affect cell survival of MCF-10A and HaCaT at the concentration of 1–2.5 to 5–10 μM after 24, 48, or 72 h of treatment. However, although significant differences in cell survival between HNC and MCF-10A and HaCaT cells were observed at the 15 μM concentration, VIO affected to some extent MCF-10A and HaCaT cell survival at this dose. The distinct VIO effect on cancer and non-tumorigenic cell lines might be due to VIO-mediated inhibition of specific signaling transduction pathways which are aberrantly activated in cancer cells.
It has been shown that the activation of caspase-8 by VIO is mediated by direct activation of the tumor necrosis factor receptor 1 (TNFR1) in leukemia cells and that HNC cell lines express TNFR1 [7, 41]. Thus, the cleavage of caspase-8 in HNC cells upon VIO treatment may be mediated by the activation of TNFR1. It has been shown that treatment with VIO induces the production of ROS in colon carcinoma and Ehrlich’s ascites cell lines [10–12]. We demonstrated that VIO treatment of HNC cells induces an increase in ROS production. ROS are known to trigger the intrinsic apoptotic cascade via interactions with proteins of the mitochondrial permeability transition complex [42, 43]. The p53 protein activates DNA repair, cell cycle arrest, or apoptosis in response to DNA damage [44]. In the HNC cell lines analyzed, treatment with VIO reduced the expression of p53, with the appearance of a low molecular weight product, indicative of a degradation process. Accordingly, p53, after the activation of the intrinsic pathways of apoptosis, may be ubiquitinated and degraded.
During autophagy, the ubiquitin-like LC3 protein or ATG8 is converted to LC3-I and LC3-II, the latter being a specific marker for the formation of the autophagosome [30]. Treatment with VIO was able to induce increased expression of LC3-I and LC3-II in all cell lines analyzed, thus suggesting a possible role for autophagy in the inhibition of HNC cell growth. Bcl-2 is a key regulator of apoptosis and autophagy. It has been shown that Bcl-2 prevents apoptosis by inhibiting pro-apoptotic proteins, as Bax, and inhibits autophagy by binding to Beclin1 [45]. Beclin1 is a Bcl-2-interacting protein that promotes autophagy and is inhibited by binding with Bcl-2. Thus, the decreased expression of Bcl-2 upon VIO treatment in HNC cell lines could make Beclin1 available to promote autophagy. To our knowledge, this is the first report demonstrating VIO-mediated autophagy in cancer cells.
Previous studies showed that HNC cell lines overexpress the ErbB receptor family, which in turn plays an essential role in the mechanisms of carcinogenesis of head and neck tumors [46]. The MAP kinases ERK1/2 are involved in the transduction pathways triggered by ErbB receptors. Their role as survival factors in several tumors is associated with their ability to stimulate nuclear translocation and activation of the transcription factor NF-κB [47]. The NF-κB pathway is often aberrantly activated during the development and progression of HNC [47, 48]. Previous studies have shown that VIO, while having no effect on the MAPK pathway, reduces the activity of NF-κB in colorectal cancer cell lines or activates NF-κB in leukemia cells through the activation of TNFR1 [7, 13]. Our results demonstrated that VIO treatment was able to inhibit ERK1/2 phosphorylation and NF-κB nuclear translocation in HNC cells. It was shown that NF-κB is involved in inflammatory processes and in the transformation, survival, and invasiveness of HNC and that the ability of different chemotherapeutic compounds to inhibit NF-κB activation is the basis of the therapeutic effects of these compounds [49–51]. The VIO-mediated inhibition of ERK1/2 may be responsible for the significant accumulation of NF-κB in the cytoplasm and may induce cell death and the inhibition of cell proliferation. In addition, the activation of NF-κB induced the expression of the anti-apoptotic Bcl-2 family genes [52]. Thus, NF-κB inhibition may initiate the apoptotic and autophagic processes via Bcl-2 downregulation.
It has been shown that intraperitoneally administered VIO showed anti-proliferative activity against subcutaneous transplanted HNC in nude mice in vivo [53].
Because a limited number of animal studies are available on the anti-cancer effects of VIO, we evaluated the in vivo effects of intratumoral administration of VIO in hindering the growth of transplanted Neu-overexpressing BALB-neuT SALTO in BALB/c mice.
We demonstrated that VIO induced SALTO apoptosis, cell growth inhibition, and inhibition of ERK1/2 activation in vitro. Then, we performed an in vivo study. VIO was intratumorally administered 3 weeks after a SALTO tumor cell challenge. Our results demonstrated that VIO was able to significantly delay tumor growth and prolong median survival in agreement with our in vitro observations. In addition, it has been reported that the administration of VIO was safe, since no alteration of hematology and biochemistry parameters (ALT, AST, and creatinine levels) has been observed in BALB/c mice treated with daily i.p. doses of VIO up to 1 mg/kg for 35 days. Furthermore, VIO did not cause hematotoxicity, renal, and hepatotoxicity in mice [11]. Few other papers have intraperitoneally administered VIO in vivo [11, 53]. This is the first time that VIO has been intratumorally administered. Our findings may play a significant role in planning cancer therapy protocols using VIO or other bacteria derivatives for the treatment of salivary gland tumors and other accessible tumors by intratumoral administration.
Taken together, our results indicate that the treatment of HNC cells with VIO may be useful in the inhibition of in vivo and in vitro cancer cell growth. Because the molecular mechanisms of the action of VIO involve the activation of specific signaling transduction pathways, which vary depending on the cell type, additional studies performed both in vitro and in vivo will be necessary to fully define the anti-cancer therapeutic potential of this compound.
References
Matz C, Deines P, Boenigk J, Arndt H, Eberl L, Kjelleberg S, et al. Impact of violacein-producing bacteria on survival and feeding of bacteriovorans nanoflagellates. Appl Environ Microbiol. 2004;70:1593–9.
Johnson JH, Tymiak AA, Bolgar MS. Janthinocins A, B and C novel peptide lactone antibiotics produced by Janthinobacterium lividum. II. Structure elucidation. J Antibiot. 1990;43:920–30.
O'sullivan J, McCullough J, Johnson JH, Bonner DP, Clark JC, Dean L, et al. Janthinocins A, B and C, novel peptide lactone antibiotics produced by Janthinobacterium lividum. I. Taxonomy, fermentation, isolation, physico-chemical and biological characterization. J Antibiot. 1990;4:913–9.
Leon L, Miranda CC, De Souza AO, Dùran N. Antileishmanial activity of the violacein extracted from Chromobacterium violaceum. J Antimicrob Chemother. 2001;4:449–50.
Andrighetti-Fronher CR, Antonio RV, Creczynsky-Pasa TB, Barardi CR, Simoes CM. Cytotoxicity and potential antiviral evaluation of violacein produced by Chromobacterium violaceum. Mem Inst Oswaldo Cruz. 2003;98:843–8.
Dessaux Y, Elmerich C, Faure D. Violacein: a molecule of biological interest originating from the soil-borne bacterium Chromobacterium violaceum. Rev Med Interne. 2004;25:659–62.
Ferreira CV, Bos CL, Versteeg HH, Justo GZ, Duran N, Peppelenbosch MP. Molecular mechanism of violacein-mediated human leukemia cell death. Blood. 2004;104:1459–64.
Ballestriero F, Daim M, Penesyan A, Nappi J, Schleheck D, Di Bazzicalupo P, et al. Antinematode activity of violacein and the role of the insulin/IGF-1 pathway in controlling violacein sensitivity in Caenorhabditis elegans. PLoS ONE. 2014;9(10):e109201.
Pantanella F, Berlutti F, Passariello C, Sarli S, Morea C, Schippa S. Violacein and biofilm production in Janthinobacterium Lividum. J Appl Microbiol. 2007;102:992–9.
Durán N, Justo GZ, Ferreira CV, Melo PS, Cordi L, Martins D. Violacein: properties and biological activities. Biotechnol Appl Biochem. 2007;48:127–33.
Bromberg N, Dreyfuss JL, Regatieri CV, Palladino MV, Durán N, Nader HB, et al. Growth inhibition and pro-apoptotic activity of violacein in Ehrlich ascites tumor. Chem Biol Interact. 2010;186:43–52.
de Carvalho DD, Costa FT, Duran N, Haun M. Cytotoxic activity of violacein in human colon cancer cells. Toxicol In Vitro. 2006;20:1514–21.
Kodach LL, Bos CL, Durán N, Peppelenbosch MP, Ferreira CV, Hardwick JC. Violacein synergistically increases 5-fluorouracil cytotoxicity, induces apoptosis and inhibits Akt-mediated signal transduction in human colorectal cancer cells. Carcinogenesis. 2006;27:508–16.
Platt D, Amara S, Mehta T, Vercuyssee K, Myles EL, Johnson T, et al. Violacein inhibits matrix metalloproteinase mediated CXCR4 expression: potential anti-tumor effect in cancer invasion and metastasis. Biochem Biophys Res Commun. 2014;455:107–12.
Wilken R, Veena MS, Wang MB, Srivatsan ES. Curcumin: a review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol Cancer. 2011;10:12–31.
Rothenberg SM, Ellisen LW. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest. 2012;122:1951–7.
Matta A, Ralhan R. Overview of current and future biologically based targeted therapies in head and neck squamous cell carcinoma. Head Neck Oncol. 2009;1:6–14.
Theisen C. Chemoprevention: what’s in a name? J Natl Cancer Inst. 2001;93:743.
Menezes CB, Silva BP, Sousa IM, Ruiz AL, Spindola HM, Cabral E, et al. In vitro and in vivo antitumor activity of crude extracts obtained from Brazilian Chromobacterium sp isolates. Braz J Med Biol Res. 2013;46:65–70.
Newman DJ, Cragg GM. Microbial antitumor drugs: natural products of microbial origin as anticancer agents. Curr Opin Investig Drugs. 2009;10:1280–96.
Wille G, Steglich W. A short synthesis of the bacterial pigments violacein and deoxyviolacein. Synthesis. 2001;5:759–62.
Pannellini T, Spadaro M, Di Carlo E, Ambrosino E, Iezzi M, Amici A, et al. Timely DNA vaccine combined with systemic IL-12 prevents parotid carcinomas before a dominant-negative p53 makes their growth independent of HER-2/neu expression. J Immunol. 2006;176:7695–703.
Masuelli L, Marzocchella L, Quaranta A, Palumbo C, Pompa G, Izzi V, et al. Apigenin induces apoptosis and impairs head and neck carcinomas EGFR/ErbB2 signaling. Front Biosci. 2011;16:1060–8.
Masuelli L, Benvenuto M, Fantini M, Marzocchella L, Sacchetti P, Di Stefano E, et al. Curcumin induces apoptosis in breast cancer cell lines and delays the growth of mammary tumors in neu transgenic mice. J Biol Regul Homeost Agents. 2013;27:105–19.
Elmann A, Mordechay S, Erlank H, Telerman A, Rindner M, Ofir R. Anti-neuroinflammatory effects of the extract of Achillea fragrantissima. BMC Complement Altern Med. 2011;11:98–108.
Masuelli L, Budillon A, Marzocchella L, Mrozek MA, Vitolo D, Di Gennaro E, et al. Caveolin-1 overexpression is associated with simultaneous abnormal expression of the E-cadherin/α-β catenins complex and multiple ErbB receptors and with lymph nodes metastasis in head and neck squamous cell carcinomas. J Cell Physiol. 2012;227:3344–53.
Masuelli L, Di Stefano E, Fantini M, Mattera R, Benvenuto M, Marzocchella L, et al. Resveratrol potentiates the in vitro and in vivo anti-tumoral effects of curcumin in head and neck carcinomas. Oncotarget. 2014;5:10745–62.
Masuelli L, Fantini M, Benvenuto M, Sacchetti P, Giganti MG, Tresoldi I, et al. Intratumoral delivery of recombinant vaccinia virus encoding for ErbB2/Neu inhibits the growth of salivary gland carcinoma cells. J Transl Med. 2014;12:122.
D’Amato V, Rosa R, D’Amato C, Formisano L, Marciano R, Nappi L, et al. The dual PI3K/mTOR inhibitor PKI-587 enhances sensitivity to cetuximab in EGFR-resistant human head and neck cancer models. Br J Cancer. 2014;110:2887–95.
Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008;445:29–76.
Chakrabarty AM. Microorganisms and cancer: quest for a therapy. J Bacteriol. 2003;185:2683–6.
Bernardes N, Seruca R, Chakrabarty AM, Fialho AM. Microbial-based therapy of cancer. Current progress and future prospects. Bioeng Bugs. 2010;1:178–90.
Dufosse L. Pigments, microbial. Encyclopedia Microbiol. 2009;4:457–71.
Nemunaitis J, Khuri F, Ganly I, Arseneau J, Posner M, Vokes E, et al. Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer. J Clin Oncol. 2001;19:289–98.
Masuelli L, Marzocchella L, Focaccetti C, Lista F, Nardi A, Scardino A, et al. Local delivery of recombinant vaccinia virus encoding for neu counteracts growth of mammary tumors more efficiently than systemic delivery in neu transgenic mice. Cancer Immunol Immunother. 2010;59:1247–58.
Galanis E, Russell S. Cancer gene therapy clinical trials: lessons for the future. Br J Cancer. 2001;85:1432–6.
Forsyth P, Roldán G, George D, Wallace C, Palmer CA, Morris D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16:627–32.
Roberts NJ, Zhang L, Janku F, Collins A, Bai RY, Staedtke V, et al. Intratumoral injection of Clostridium novyi-NT spores induces antitumor responses. Sci Transl Med. 2014;6:249ra111.
Fujiwara S, Wada H, Miyata H, Kawada J, Kawabata R, Nishikawa H, et al. Clinical trial of the intratumoral administration of labeled DC combined with systemic chemotherapy for esophageal cancer. J Immunother. 2012;35:513–21.
Queiroz KC, Milani R, Ruela-de-Sousa RR, Fuhler GM, Justo GZ, Zambuzzi WF, et al. Violacein induces death of resistant leukaemia cells via kinome reprogramming, endoplasmic reticulum stress and Golgi apparatus collapse. PLoS ONE. 2012;7(10):e45362.
Jackson-Bernitsas DG, Ichikawa H, Takada Y, Myers JN, Lin XL, Darnay BG, et al. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene. 2007;26:1385–97.
Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–40.
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems and apoptosis. Free Radic Biol Med. 2010;48:749–62.
Levine AJ, Hu W, Feng Z. The p53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–36.
Lian J, Karnak D, Xu L. The Bcl-2-Beclin interaction in (−)-gossypol-induced autophagy versus apoptosis in prostate cancer cells. Autophagy. 2010;6:1201–3.
Bei R, Pompa G, Vitolo D, Moriconi E, Ciocci L, Quaranta M, et al. Colocalization of multiple ErbB receptors in stratified epithelium of oral squamous cell carcinoma. J Pathol. 2001;195:343–8.
Vanden Berghe W, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, et al. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–90.
Ondrey F, Dong G, Sunwoo J, Chen Z, Wolf JS, Crowl-Bancroft CV, et al. Constitutive activation of transcription factors NF-kB, AP-1 and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express proinflammatory and proangiogenic cytokines. Mol Carcinog. 1999;26:119–29.
Caicedo-Granados EE, Wuertz BR, Marker PH, Lee GS, Ondrey FG. The effect of indomethacin on paclitaxel sensitivity and apoptosis in oral squamous carcinoma cells: the role of nuclear factor-κB inhibition. Arch Otolaryngol Head Neck Surg. 2011;137:799–805.
Allen CT, Ricker JL, Chen Z, Van Waes C. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 2007;29:959–71.
Van Waes C, Chang AA, Lebowitz PF, Druzgal CH, Chen Z, Elsayed YA, et al. Inhibition of nuclear factor-kappaB and target genes during combined therapy with proteasome inhibitor bortezomib and reirradiation in patients with recurrent head-and-neck squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2005;63:1400–12.
Bui NT, Livolsi A, Peyron JF, Prehn JH. Activation of nuclear factor kappaB and Bcl-x survival gene expression by nerve growth factor requires tyrosine phosphorylation of IkappaBalpha. J Cell Biol. 2001;152:753–64.
Hashimi SM, Xu T, Wei MQ. Violacein anticancer activity is enhanced under hypoxia. Oncol Rep. 2015;33:1731–6.
Acknowledgments
This research was supported by Sapienza Università di Roma grant nos. C26A12AXEH and C26A138TC2 (LM), grant no. C26A14TLFT (GLR), and Bando Futuro in Ricerca 2010 grant no. RBFR10ZJQT (GLR). The authors thank Mrs. Lucilla Simonelli for the technical assistance.
Rosanna Mattera is recipient of the Sapienza PhD program in Moleculare Medicine.
Enrica Di Stefano is recipient of the Sapienza PhD program in Biotechnology in Clinical Medicine.
Author information
Authors and Affiliations
Corresponding author
Additional information
Laura Masuelli and Fabrizio Pantanella contributed equally to this work.
Rights and permissions
About this article

Cite this article
Masuelli, L., Pantanella, F., La Regina, G. et al. Violacein, an indole-derived purple-colored natural pigment produced by Janthinobacterium lividum, inhibits the growth of head and neck carcinoma cell lines both in vitro and in vivo. Tumor Biol. 37, 3705–3717 (2016). https://doi.org/10.1007/s13277-015-4207-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4207-3