
Abstract
Despite the recent advances in diagnostic and therapeutic strategies, oral squamous cell carcinoma (OSCC) remains a major health burden. Protein biomarker discovery for early detection will help to improve patient survival rate in OSCC. Mass spectrometry-based proteomics has emerged as an excellent approach for detection of protein biomarkers in various types of cancers. In the current study, we have used 4-Plex isobaric tags for relative and absolute quantitation (iTRAQ)-based shotgun quantitative proteomic approach to identify proteins that are differentially expressed in cancerous tissues compared to normal tissues. The high-resolution mass spectrometric analysis resulted in identifying 2,074 proteins, among which 288 proteins were differentially expressed. Further, it was noticed that 162 proteins were upregulated, while 125 proteins were downregulated in OSCC-derived cancer tissue samples as compared to the adjacent normal tissues. We identified some of the known molecules which were reported earlier in OSCC such as MMP-9 (8.4-fold), ZNF142 (5.6-fold), and S100A7 (3.5-fold). Apart from this, we have also identified some novel signature proteins which have not been reported earlier in OSCC including ras-related protein Rab-2A isoform, RAB2A (4.6-fold), and peroxiredoxin-1, PRDX1 (2.2-fold). The immunohistochemistry-based validation using tissue microarray slides in OSCC revealed overexpression of the RAB2A and PRDX1 gene in 80 and 68 % of the tested clinical cases, respectively. This study will not only serve as a resource of candidate biomarkers but will contribute towards the existing knowledge on the role of the candidate molecules towards disease progression and therapeutic potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Head and neck cancer is the sixth most common cancer worldwide. Half of these cases comprise of squamous cell carcinoma within the oral cavity or on the lip [1]. The progress of oral cancer is associated with multistep biological events, and the key threats to its association with the advancement of OSCC are alcohol consumption, tobacco [2], genetic mutation, tobacco-free products, and human papilloma virus (HPV) infections [3–5]. Global scenario shows that every year around 600,000 peoples are affected by head and neck cancer. Among this population, survival of 40 % cases is less than 5 years from the initial time of disease progression [6]. The clinical opinion of the OSCC is mostly indigent. Owing to delayed disease appearance of the patients and lack of appropriate molecules to identify the development of illness, there is an urgent need to find early detection biomarkers for oral cancer for predicting survival, diagnosis, and overall prognosis. Proteomics is an advanced approach for the detection of protein biomarker with altered levels which may help in an early detection of protein markers for diagnostic purposes [7]. The literature survey noticeably indicates that further additional research work has to be done in depth through proteomic approach so that OSCC proteome would lead to biomarker discovery. Proteomics has been effectively engaged in the study of breast, lung, gastrointestinal, prostate, and head and neck squamous cell carcinoma (HNSCC) [8, 9]. In the present study, we have used high-resolution mass spectrometry combined with isobaric tags for relative and absolute quantitation (iTRAQ) labeling technology for the characterization and quantitation of the proteins in cancerous and non-cancerous oral tissue. The labeling method permits multiplexing as well as relative quantitation of various proteins by the addition of chemical mass tags. It has been observed that characteristic features of the tagged peptides or proteins will not affect any structural, analytical, or biochemical changes by the present labeling strategy [9]. Using this mass spectrometry approach, our findings resulted in identification of 2,074 proteins, out of which 288 proteins were differentially expressed in OSCC-derived cancerous tissue samples as compared to the normal adjacent tissues. This study led to the identification of known molecules as well as novel molecules which have not been reported in OSCC confirming the effectiveness of multiplexing for cancer biomarker discovery. Further confirmation in large number of oral cancer populations can give a direction towards smooth and highly propitious clinical and scientific tool for OSCC diagnosis or monitoring the cancer progression of patients.
Materials and methods
Tissue sample collection
The current research work was approved by the institutional human ethics committee [approval number: PR-HC/6-119/2895(8)] at the Bankura Sammilani Medical College, Bankura, West Bengal. Tumor tissue samples and adjacent normal (3–4 cm away from tumor site) tissue were collected from ten OSCC patients. The clinical details of the patients are given in the Supplementary Table. The patient informed consent was signed by all individual participants prior to the study for the collection of samples. All samples were histologically graded and reviewed by an expert pathologist. Samples were stored at −80 °C in a freezer.
Protein isolation, iTRAQ labeling, and SCX fractionation
About 10 mg equivalents of OSCC and normal tissue were homogenized and sonicated using 0.5 % SDS with a cell disperser. The cell debris was eliminated by centrifugation at 12,000 rpm for 30 min at 4 °C, and supernatant was collected and the protein concentration was determined by Lowry’s method. The trypsin digestion and labeling were carried out with 4-Plex iTRAQ reagents (iTRAQ Reagents Multiplex kit; Applied Biosystems/MDS Sciex, CA, USA) as depicted earlier [8]. The tryptic digests were labeled with iTRAQ reagents as follows: peptides derived from pooled normal were labeled with a reagent containing reporter ions 114 and 115 whereas from cancerous tissues were labeled with reporter ions 116 and 117 in technical replicates. Later, the samples were pooled and vacuum dried to reduce the sample volume to 100 μl in the post-labeling stage. The pooled mixture of peptides was fractionated using strong cation exchange (SCX) chromatography as previously described [8]. An aggregate of 96 fractions was collected in a 96-well plate, and further least complex fractions were pooled based on the peak intensity (optical density) as per the chromatogram and make it to 21 best fractions. Pooled fractions were collected to be vacuum dried and subsequently desalted using C18 STAGE tips and stored at −20 °C till further analysis.
Mass spectrometry analysis
SCX fractions were analyzed on an LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific, Bremen, Germany) interfaced with Proxeon Easy nLC system (Thermo Scientific, Bremen, Germany) as mentioned previously [10]. Briefly, the mass spectrometry (MS) and MS/MS scans were acquired in an Orbitrap mass analyzer at a mass resolution of 60,000 and 15,000 at 400 m/z, respectively. Full MS scans were acquired in the m/z range of 350–1,800. In each duty cycle, 20 most abundant precursor ions with charge state ≥2 were sequentially isolated to a target value of 50,000 ions for fragmentation using higher energy collision dissociation (HCD) mode at 41 % normalized collision energy. Isolation width was set to 2 m/z singly charged precursor ions, and precursors with unassigned charge states were rejected. The acquired ions were dynamically excluded for 45 s. The automatic gain control for full MS and MS/MS was set to 1 × 106 and 5 × 104 ions, respectively. The maximum ion accumulation time was set to 200 ms for MS and 300 ms for MS/MS scans. The lock mass option was enabled using polysiloxane ion (m/z, 445.120025) from ambient air for internal calibration.
Protein expression by immunoblot analysis
Tissue immunoblot analysis was performed to evaluate the expression profile of the Ras-related protein Rab-2A isoform a (RAB2A) and peroxiredoxin-1 (PRDX1). Tissue samples were lysed and subjected to immunoblot analysis according to an earlier reported method [11]. The intensity of the protein bands was analyzed by densitometry, after normalization to the corresponding protein controls.
Immunohistochemistry
Immunohistochemistry (IHC) was performed by using tissue arrays purchased from US Biomax as previously explained [11]. Briefly, all the array slides were examined and evaluated by two independent pathologists based on the intensity of staining and scored as negative (0), mild (1+), moderate (2+), and strong (3+). The distribution of staining of cancer cells was scored as 0 (less than 5 % of cell staining), 1+ (5–30 % of cell staining), 2+ (31–60 % of cell staining), and 3+ (greater than 60 % of cell staining).
Statistical analysis
Statistical analysis was carried out with GraphPad Prism version 5.0. The relationship between RAB2A and PRDX1 expression levels and clinicopathological parameters were analyzed using chi-square and Student’s t test. In all tests, two-sided p values <0.05 were considered statistically significant.
Results
Quantitative mass spectrometric analysis revealed altered profile of number proteins in OSCC
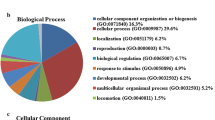
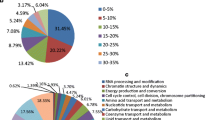
Quantitative mass spectrometric analysis of cancerous and non-cancerous tissue was conducted by LC-MS/MS using LTQ-Orbitrap Velos, which resulted in the identification of 2,074 proteins from 21 fractions. The tissue proteome analysis approach being used is summarized in Fig. 1. Apart from this, the entire list of proteins, including a peptide sequence, m/z values, a charge state, and iTRAQ ratios, is provided as Supplementary Tables 1 and 2. Based on iTRAQ quantification, we found 288 proteins to be differentially expressed in cancerous tissue with comparison to non-cancerous tissue. Among these, 126 proteins were found to be overexpressed (<2-fold), and 162 were found to be downregulated (>2-fold) in the tissue proteome. The partial list of known molecules and those identified which were novel but have not been studied in OSCC are reported in Tables 1 [12–21] and 2, respectively. The representative MS/MS spectra of the two molecules RAB2A and PRDX1 have been shown in subpanels a and b of Fig. 2, respectively. The insets in the figure illustrate the spectra of the individual peptide of that particular protein with relative abundance of iTRAQ reporter ions. The OSCC tissue proteome information was explored with the help of Human Protein Reference Database (http://www.hprd.org) which also included manually investigated information from published literature [22]. "Gene ontology (GO)" annotation was performed by mapping the proteins identified in OSCC proteome to HPRD database which was fulfilled with GO conditions for functional explanation of proteins. The bioinformatic analysis was carried out to categorize identified proteins based on subcellular localization and biological function, which is represented in subpanels c and d of Fig. 2, respectively. The summary also features fold changes for differentially expressed proteins in OSCC. Here, 863 proteins were identified with a single peptide; 363 were with two peptides and 849 were identified with more than two peptides. Around 23 % of the proteins were found to be membranous or extracellular protein and remaining proteins for which expression was found to be in ribosome, nucleus, mitochondria, and endoplasmic reticulum and the cytoplasm.

Workflow implemented for OSCC tissue proteome analysis by iTRAQ labeling methodology was engaged to quantitate the abundance of proteins in cancerous and non-cancerous tissue. Proteins were isolated and subjected to trypsin digestion followed by iTRAQ labeling. Post-labeling the equal amounts of proteins from tissue lysate was iTRAQ labeled from pooled cancerous and non-cancerous tissue followed by strong cation exchange chromatography. The fractions were subjected to LC-MS/MS analysis on a LTQ-Orbitrap Velos mass spectrometer. The data were analyzed and quantitated using Proteome Discoverer software suite. Some of the novel upregulated proteins were validated using IHC and Western blot

MS and MS/MS spectra of peptide from representative differentially expressed proteins identified in this study. a RAB2A. b PRDX1. Classification of proteins by gene ontology based on their cellular localization and biological process. c Distribution of proteins based on their biological process using gene ontology classifier. d Distribution of proteins based on their cellular component using gene ontology classifier
Immunoblot analysis provided the validation of iTRAQ findings in OSCC
To authenticate the iTRAQ results obtained from the from proteomic data, RAB2A and PRDX1 expression was evaluated in the cancerous and non-cancerous oral tissue lysate by using immunoblot analysis. We observed the overexpression of both the proteins which was in concordance with the LC-MS/MS results, as shown in Fig. 3.

Validation of iTRAQ results was done by Western blot analysis of PRDX1 and RAB2A in the cancerous and non-cancerous oral tissue specimens. The samples were resolved by SDS-PAGE, subsequently electroblotted on to nitrocellulose membrane, and probed with specific antibodies. β-actin was used as a loading control. T = tumor tissues, N = normal tissues. Data were presented as mean ± SD, ***p < 0.05, Student’s t test
Validation of novel proteins by immunohistochemical analysis in OSCC
In order to study the clinical and biological significance of the recognized biomarkers, tissue microarray-based validation of RAB2A and PRDX1 proteins was carried out by IHC. In the present study, RAB2A is mostly cytoplasmic, and expression was observed in 80 % of the OSCC cases (45/56) as shown in Fig. 4a. Based on the staining intensity, cases were classified into four sub-categories—11/56 (19.64 %) cancers with negative expression, 7/45 (15.55 %) with mild (1+) expression, 8/45 (17.77 %) with moderate (2+) expression, and 30/45 (66 %) with strong (3+) expression. Apart from this, normal adjacent oral tissue was stained and, compared with the reference for RAB2A expression, it shows 4/10 (40 %) absent or very mild expression in the basal epithelium. Further, the overexpression of PRDX1 was also found in OSCC patients, and it is generally localized in membranous and cytoplasmic region with granular staining. In the current study, PRDX1 expression was found to be overexpressed in 132/180 (73 %) of the OSCC cases. Further, the staining intensity was measured and classified into four groups such as cancers with negative (0), mild (1+), moderate (2+), and strong (3+) expression, which were 48/180 (26.66 %), 28/132 (21.21 %), 18/132 (13.63 %), and 96/132 (72.72 %), respectively (shown in Fig. 4b). Summary of immunohistochemical labeling and immune reactivity scoring (IRS) as per the staining intensity of different molecules in OSCC cases have been shown in Fig. 4c, d. Here, we came to recognize that RAB2A and PRDX1 expression in cancer tissue compared with normal tissue has been increased significantly (p < 0.001). Furthermore, we evaluated the relationship between RAB2A and PRDX1 expression in OSCC samples as per the scoring pattern. For both the proteins, mild (1+) versus moderate (2+) scoring pattern, p value was not statistically significant. Apart from this, all the scoring patterns were statistically significant (p < 0.001) as shown in Fig. 4e. Hence, this data has importance in the clinicopathological aspect of the early detection and prevention of cancer.

Validation using immunohistochemical staining of RAB2A and PRDX1 in cancerous and non-cancerous tissues using tissue microarrays. Representative sections at the magnification of ×20 from tissue microarrays stained with anti-RAB2A and anti-PRDX1 show a expression pattern of RAB2A in representative normal oral squamous tissue and oral squamous cell carcinoma (OSCC) and b expression pattern of PRDX1 in normal oral squamous tissue and oral squamous cell carcinoma (OSCC). Staining pattern, p value, and cellular localization of Ras-related protein (RAB2A) and Peroxiredoxin-1 (PRDX1). c The IRS staining scores of RAB2A and PRDX1 in the normal vs tumor-positive tumor sample were evaluated (n = 3). d Summary of immunohistochemical labeling of different molecules in OSCC cases. e Statistical correlation of RAB2A and PRDX1 data was presented as mean ± SD. *p value is not statistically significant, p < 0.001, by chi-square test
Discussion
In recent years, the major scientific progress in the field of proteomics has been given a scope in recognition of proteins from various cancer categories. Quantitative proteomic approach has made possible to know multiple potential biomarkers. The present research is based on a tissue proteome investigation of oral tissue squamous cell carcinoma. This study led to the identification of 287 differentially expressed proteins. Many of them are novel molecules that are highly expressed in several human cancer, and it is not been explained in oral squamous cell carcinoma earlier. Corneodesmosin precursor is generally sited in the major histocompatibility complex (MHC) class I region on chromosome number 6. It is expressed in epithelial cells in the infiltrative aggressive relapsing tumor of basal cell carcinoma [23]. In our current experiment, we have found more than 2-fold increase in the expression of corneodesmosin with comparison to the normal. Another molecule Secerinin-1 which showed some high expression (2-fold up) is a cytosolic protein that excites exocytosis in mast cells, but unfortunately, the mechanisms of its function in exocytosis remain unexplored [24]. Apart from this, Lumican has an important role in regulating cell behavior, embryonic development, tissue repair, and cancerous growth [25]. The invasive cell migration was also being noticed in many cancers. The upregulation was also being observed in pancreatic, colorectal, breast, and uterine cervical cancers [26–29]. Another group of protein Ras-related protein 14 (RAB14) showed some significance and fold change. Its overexpression has been reported earlier in non-small cell lung carcinoma [30]. Among upregulated proteins, RAB2A (4.6-fold) is one of the novel biomarker identified and validated using OSCC TMAs in the current study. Rab family that are small molecular weight membrane-bound proteins, which are guanosine triphosphatases (GTPases), are involved in vesicular fusion and trafficking. It has highly extremely conserved domains which are responsible for GTP binding and hydrolysis. This protein is derived from pre-Golgi intermediates, and its primary function is protein transport from the endoplasmic reticulum (ER) to the Golgi complex [31]. It is crucial for vesicular transport from the Golgi to the nuclear envelope in spermatids during acrosomal biogenesis [32]. It is a membranous protein that is involved in vesicular fusion and organelle biogenesis. Further abnormality in RAB function leads to cancer. We have also identified PRDX1 that is expressed to be in higher abundance (2.2-fold) in the OSCC tissue lysate. PRDX1 is a thiol-specific peroxidase that decreases and detoxifies a broad range of organic hydroperoxides such as H2O2 [33]. It is an antioxidant which has a serious role in cancer progression and plays a significant role in the growth and development of several human cancers and influences in various cellular processes including cell survival, proliferation, and apoptosis. The biological function of PRDX1 in cancer is still ambiguous, and its mechanism has not been elucidated so far. Previous studies have shown that PRDX1 functions as a tumor suppressor in several types of cancers, but other studies have indicated that it is overexpressed in some types of human cancers [34]. PRDX1 has been studied in different types of cancer progression and overexpression has been detected in pulmonary and thyroid carcinomas, and the exact function and mechanisms of PRDX1 upregulation in colon cancer have also been reported [35, 36]. Interestingly, transgenic mice deficient of PRDX1 gene developed several cancers at higher incidence [37]. It has now been noticed that PRDX1 has a regulatory function which can cause cell survival, cell proliferation, cell death, metabolism, and DNA repair. PRDX1 also had a role in suppressing radiation-induced c-Jun N-terminal kinase (JNK) activation and further leads to apoptosis in human lung cancer cells through interaction with the GSTpi-JNK complex, which does not involve antioxidant activity [38].
In conclusion, the current research presented identification and validation of RAB2A and PRDX1 by LC-MS/MS, immunoblot, and immunohistochemistry-based proteomic approach. Thus, tissue proteomic analysis will help as a resource of novel biomarker development in the field of cancer biomarker discovery. Though in vitro findings cannot fully represent in vivo conditions, an in vitro study can be used as an initial phase for confirmation. Using sophisticated mass spectrometry-based methods like multiple reactions monitoring, these potential biomarkers can be further validated on a large number of patient samples. The specificity and sensitivity of these biomarkers need to be translated in clinical research, and this will further help to find probable strategies for the oral cancer biomarker development which may lead to improved patient outcomes.
References
Kreeft AM, Tan IB, Leemans CR, Balm AJ. The surgical dilemma in advanced oral and oropharyngeal cancer: how we do it. Clin Otolaryngol. 2011;36(3):260–6. doi:10.1111/j.1749-4486.2011.02299.x.
Ni YH, Ding L, Hu QG, Hua ZC. Potential biomarkers for oral squamous cell carcinoma: proteomics discovery and clinical validation. Proteomics: Clin Appl. 2015;9(1-2):86–97. doi:10.1002/prca.201400091.
Mangalath U, Aslam SA, Abdul Khadar AH, Francis PG, Mikacha MS, Kalathingal JH. Recent trends in prevention of oral cancer. J Int Soc Prev Community Dent. 2014;4 Suppl 3:S131–8. doi:10.4103/2231-0762.149018.
Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Reboiras-Lopez MD, Gandara Rey JM, Garcia-Garcia A. Genetic and molecular alterations associated with oral squamous cell cancer (review). Oncol Rep. 2009;22(6):1277–82.
Mishra A, Verma V. Oral sex and HPV: population based indications. Indian J Otolaryngol Head Neck Surg. 2015;67 Suppl 1:1–7. doi:10.1007/s12070-012-0521-x 521.
Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008;83(4):489–501. doi:10.4065/83.4.489.
Turhani D, Krapfenbauer K, Thurnher D, Langen H, Fountoulakis M. Identification of differentially expressed, tumor-associated proteins in oral squamous cell carcinoma by proteomic analysis. Electrophoresis. 2006;27(7):1417–23. doi:10.1002/elps.200500510.
Marimuthu A, Chavan S, Sathe G, Sahasrabuddhe NA, Srikanth SM, Renuse S, et al. Identification of head and neck squamous cell carcinoma biomarker candidates through proteomic analysis of cancer cell secretome. Biochim Biophys Acta. 2013;1834(11):2308–16. doi:10.1016/j.bbapap.2013.04.029.
Pawar H, Kashyap MK, Sahasrabuddhe NA, Renuse S, Harsha HC, Kumar P, et al. Quantitative tissue proteomics of esophageal squamous cell carcinoma for novel biomarker discovery. Cancer Biol Ther. 2011;12(6):510–22. doi:10.4161/cbt.12.6.16833.
Syed N, Chavan S, Sahasrabuddhe NA, Renuse S, Sathe G, Nanjappa V, et al. Silencing of high-mobility group box 2 (HMGB2) modulates cisplatin and 5-fluorouracil sensitivity in head and neck squamous cell carcinoma. Proteomics. 2015;15(2-3):383–93. doi:10.1002/pmic.201400338.
Parida S, Parekh A, Dey G, Ghosh SC, Mandal M. Molecular inhibition of prostaglandin E2 with GW627368X: therapeutic potential and preclinical safety assessment in mouse sarcoma model. Cancer Biol Ther. 2015. doi:10.1080/15384047.2015.1040953.
Franchi A, Santucci M, Masini E, Sardi I, Paglierani M, Gallo O. Expression of matrix metalloproteinase 1, matrix metalloproteinase 2, and matrix metalloproteinase 9 in carcinoma of the head and neck. Cancer. 2002;95(9):1902–10. doi:10.1002/cncr.10916.
Miyake N, Katoh O, Hirata S, Kimura S, Watanabe H, Yajin K. Expression of the Kruppel-type zinc finger gene, ZK7, in head and neck squamous cell carcinoma and normal mucosa. Cancer Lett. 2002;185(1):111–8.
Polachini GM, Sobral LM, Mercante AM, Paes-Leme AF, Xavier FC, Henrique T, et al. Proteomic approaches identify members of cofilin pathway involved in oral tumorigenesis. PLoS One. 2012;7(12):e50517. doi:10.1371/journal.pone.0050517.
Kaur J, Ralhan R. Differential expression of 70-kDa heat shock-protein in human oral tumorigenesis. Int J Cancer. 1995;63(6):774–9.
Kesting MR, Sudhoff H, Hasler RJ, Nieberler M, Pautke C, Wolff KD, et al. Psoriasin (S100A7) up-regulation in oral squamous cell carcinoma and its relation to clinicopathologic features. Oral Oncol. 2009;45(8):731–6. doi:10.1016/j.oraloncology.2008.11.012.
Feher LZ, Pocsay G, Krenacs L, Zvara A, Bagdi E, Pocsay R, et al. Amplification of thymosin beta 10 and AKAP13 genes in metastatic and aggressive papillary thyroid carcinomas. Pathol Oncol Res. 2012;18(2):449–58. doi:10.1007/s12253-011-9467-7.
Herold-Mende C, Andl T, Laemmler F, Reisser C, Eichhorn S. Expression and localization profile of tenascin in squamous cell carcinomas of the head and neck. HNO. 1999;47(8):723–9.
Bagutti C, Speight PM, Watt FM. Comparison of integrin, cadherin, and catenin expression in squamous cell carcinomas of the oral cavity. J Pathol. 1998;186(1):8–16. doi:10.1002/(SICI)1096-9896(199809)186.
Chiang WF, Hwang TZ, Hour TC, Wang LH, Chiu CC, Chen HR, et al. Calreticulin, an endoplasmic reticulum-resident protein, is highly expressed and essential for cell proliferation and migration in oral squamous cell carcinoma. Oral Oncol. 2013;49(6):534-–41. doi:10.1016/j.oraloncology.2013.01.003.
Gourin CG, Zhi W, Adam BL. Proteomic identification of serum biomarkers for head and neck cancer surveillance. Laryngoscope. 2009;119(7):1291–302. doi:10.1002/lary.20279.
Goel R, Muthusamy B, Pandey A, Prasad TS. Human protein reference database and human proteinpedia as discovery resources for molecular biotechnology. Mol Biotechnol. 2011;48(1):87–95. doi:10.1007/s12033-010-9336-8.
Barbaud A, Simon M, Parache RM, Serre G. Immunohistochemical characterization of the differentiation state of basal cell carcinomas with special interest for infiltrating relapsing tumors. Eur J Dermatol. 1998;8(5):320–4.
Suda T, Tsunoda T, Uchida N, Watanabe T, Hasegawa S, Satoh S, et al. Identification of secernin 1 as a novel immunotherapy target for gastric cancer using the expression profiles of cDNA microarray. Cancer Sci. 2006;97(5):411–9. doi:10.1111/j.1349-7006.2006.00194.x.
Coulson-Thomas VJ, Coulson-Thomas YM, Gesteira TF, de Andrade Paula CA, Carneiro CR, Ortiz V, et al. Lumican expression, localization and antitumor activity in prostate cancer. Exp Cell Res. 2013;319(7):967–81. doi:10.1016/j.yexcr.2013.01.023.
Ping Lu Y, Ishiwata T, Asano G. Lumican expression in alpha cells of islets in pancreas and pancreatic cancer cells. J Pathol. 2002;196(3):324–30. doi:10.1002/path.1037.
Koninger J, Giese T, di Mola FF, Wente MN, Esposito I, Bachem MG, et al. Pancreatic tumor cells influence the composition of the extracellular matrix. Biochem Biophys Res Commun. 2004;322(3):943–9. doi:10.1016/j.bbrc.2004.08.008 S0006-291X(04)01735-8.
Leygue E, Snell L, Dotzlaw H, Hole K, Hiller-Hitchcock T, Roughley PJ, et al. Expression of lumican in human breast carcinoma. Cancer Res. 1998;58(7):1348–52.
Naito Z, Ishiwata T, Kurban G, Teduka K, Kawamoto Y, Kawahara K, et al. Expression and accumulation of lumican protein in uterine cervical cancer cells at the periphery of cancer nests. Int J Oncol. 2002;20(5):943–8.
Sun J, Feng X, Gao S, Xiao Z. microRNA-338-3p functions as a tumor suppressor in human non-small-cell lung carcinoma and targets Ras-related protein 14. Mol Med Rep. 2015;11(2):1400–6. doi:10.3892/mmr.2014.2880.
Yoshida H, Miyachi M, Ouchi K, Kuwahara Y, Tsuchiya K, Iehara T, et al. Identification of COL3A1 and RAB2A as novel translocation partner genes of PLAG1 in lipoblastoma. Genes, Chromosomes Cancer. 2014;53(7):606–11. doi:10.1002/gcc.22170.
Nourashrafeddin S, Aarabi M, Modarressi MH, Rahmati M, Nouri M. The evaluation of WBP2NL-related genes expression in breast cancer. Pathol Oncol Res. 2014. doi:10.1007/s12253-014-9820-8.
Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28(1):32–40.
Gong F, Hou G, Liu H, Zhang M. Peroxiredoxin 1 promotes tumorigenesis through regulating the activity of mTOR/p70S6K pathway in esophageal squamous cell carcinoma. Med Oncol. 2015;32(2):455. doi:10.1007/s12032-014-0455-0.
Yanagawa T, Ishikawa T, Ishii T, Tabuchi K, Iwasa S, Bannai S, et al. Peroxiredoxin I expression in human thyroid tumors. Cancer Lett. 1999;145(1-2):127–32.
Kim JH, Bogner PN, Ramnath N, Park Y, Yu J, Park YM. Elevated peroxiredoxin 1, but not NF-E2-related factor 2, is an independent prognostic factor for disease recurrence and reduced survival in stage I non-small cell lung cancer. Clin Cancer Res. 2007;13(13):3875–82. doi:10.1158/1078-0432.CCR-06-2893.
Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–5. doi:10.1038/nature01819.
Kim YJ, Lee WS, Ip C, Chae HZ, Park EM, Park YM. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006;66(14):7136–42. doi:10.1158/0008-5472.CAN-05-4446.
Acknowledgements
We thank the Indian Council of Medical Research (3/2/2/207/2013/NCD-III) and the Department of Science and Technology (SR/SO/BB-58/2008) Government of India for financial support.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent
Informed consent was obtained from all individual participants included in the study.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dey, K.K., Pal, I., Bharti, R. et al. Identification of RAB2A and PRDX1 as the potential biomarkers for oral squamous cell carcinoma using mass spectrometry-based comparative proteomic approach. Tumor Biol. 36, 9829–9837 (2015). https://doi.org/10.1007/s13277-015-3758-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3758-7