
Abstract
Breast cancer is one of the most common malignancies and a major cause of cancer-related mortality all over the world. A growing body of reports revealed that microRNAs play essential roles in the progression of cancers. Aberrant expression of miR-503 has been reported in several kinds of cancer. The aim of the current study was to elucidate the role of miR-503 in the pathogenesis of breast cancer. In the present study, our results suggested that miR-503 expression was markedly downregulated in breast cancer tissues and cells. Overexpression of miR-503 in breast cancer cell lines reduced cell proliferation through inducing G0/G1 cell cycle arrest by targeting CCND1. Together, our findings provide new knowledge regarding the role of miR-503 in the progression of breast cancer and indicate the role of miR-503 as a tumor suppressor microRNA (miRNA) in breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is one of the most commonly detected cancers in women affecting about 1.2 million women worldwide each year [1]. In the past years, despite achieving significant progress in optimization of treatment with better surgery, cytotoxic agents, and endocrine therapy, the prognosis of breast cancer patients have not been altered much [2–5]. Thus, this is urgent to identify the novel molecular makers which can serve as possible therapeutic targets for treatment of breast cancer.
Recently, increasing evidences indicated that microRNAs (miRNAs), non-coding small RNAs, act as oncogenes or tumor suppressors in the development of cancer [6–8]. MiRNAs play important roles in biological progression of many types of human tumors by regulating gene expression through binding to 3′-UTR of the target mRNA [9–11]. Aberrant expression of miRNAs has been implicated in a variety of biological processes related to cancer, including proliferation, apoptosis, migration, and invasion, which act as either tumor oncogenes or suppressors [12–15]. Multiple reports indicated that miR-503 has been showed to be one of the important determinants in cancers [16–20]. However, the relationship between miR-503 and breast cancer remains incompletely understood. In the current study, we found that miR-503 was downregulated in breast cancer tissue and cell lines. Ectopic overexpression of miR-503 in breast cancer cell lines led to the inhibition of cell growth rate and cell cycle progression. Furthermore, we demonstrated that the tumor promoter gene CCND1 is a direct target of miR-503. Taken together, our results indicated that overexpression of miR-503 could inhibit cell proliferation and cell cycle progression in breast cancer by directly suppressing CCND1.
Materials and methods
Clinical specimens
Eight paired human breast cancer tissues and the adjacent non-cancerous tissues (ANT) were obtained from breast cancer patients and histopathologically diagnosed at Breast Disease Center, Department of Surgery, the First Affiliated Hospital, Sun Yat-sen University (Guangzhou, People’s Republic of China). The study was approved by the ethics committee of the First Affiliated Hospital, Sun Yat-sen University (Guangzhou, People’s Republic of China). Written informed consent was obtained from all patients. Tissue samples were collected at surgery, immediately frozen in liquid nitrogen, and stored until total RNAs or proteins were extracted.
Cell culture
Human breast cancer cell lines MCF-7, T47D, MDA-MB-231, BT549, SKBR3, ZR-75-30, and non-malignant breast epithelial cell MCF-3A were purchased from National Rodent Laboratory Animal Resource (Shanghai, People’s Republic of China) and were grown and cultured in RPMI-1640 (Gibco, USA) supplemented with 10 % fetal bovine serum (FBS, Sigma, St Louis, MI), 100 units/ml of penicillin-streptomycin (Invitrogen, Carlsbad, CA). All cells were cultured in a humidified incubator at 37 °C in an atmosphere of 95 % air and 5 % CO2.
Plasmids and transfection
MiR-503 mimic, miR-503 inhibitor, and negative control were purchased from GeneCopoeia (Guangzhou, China). MCF-7 and MDA-MB-231 cells were seeded into six-well plates 24 h before transfection to ensure 60–70 % confluence at the time of transfection. The Lipofectamine 2000 Reagent (Invitrogen, USA) was used for transfections following the manufacturer’s instructions.
RNA extraction and real-time quantitative PCR
MircoRNAs were extracted from cultured cells or patient samples with RNAiso kit for small RNA (Takara, China) according to the manufacturer’s instructions and reversely transcribed into cDNA with the One Step PrimeScript miRNA cDNA Synthesis kit (Takara, China). The resulting cDNAs were quantified with SYBR Green (Takara, China) using an ABI 7500 Fast Real-time PCR System (Applied Biosystems, USA). The relative miR-503 expression levels after normalization to U6 small nuclear RNA were calculated using 2−[(Ct of miR-503) − (Ct of U6)].
Western blotting
All the cells were gathered and lysed in cell lysis buffer 48 h after the transfection. Equal quantities of protein were separated by 10 % SDS-polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5 % non-fat milk for 2 h and incubated overnight with primary antibodies including rabbit polyclonal anti-CCND1 (1:1000; Cell Signaling Technology), anti-p21 (1:1000; Cell Signaling Technology), anti-phosphorylated retinoblastoma (anti-p-Rb, 1:1000; Cell Signaling Technology) and anti-pRb (1:1000; Cell Signaling Technology), and anti-β-actin (1:1000; Cell Signaling Technology). After being washed in TBS-T three times, PVDF membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody at a 1:5000 dilution for 2 h. Immunocomplexes were visualized using the chemiluminescence (GE, USA) following the manufacturer’s protocol.
MTT assay
MCF-7 and MDA-MB-231 cells were seeded in 96-well plates with 3 × 103 cells/well 24 h prior to the transfection of miR-503 or miR-503-in or relative control mimics (NC) and assayed 1, 2, 3, 4, 5, and 6 days after transfection. One hundred microliters 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (0.5 mg/ml, Sigma, St Louis, MI) were added into each well and the incubation continued at 37 °C for 4 h, and then, supernatant was removed and 150 μl dimethyl sulfoxide (DMSO) was added to each well to dissolve the precipitate. The absorbance at 490 nm was measured in a Thermo Scientific Multiskan (Thermo Fisher Scientific, USA).
Colony formation assay
MCF-7 and MDA-MB-231 cells were plated six-well plates at a density of 500 cells per well and incubated for 14 days. Colonies were fixed with 10 % formaldehyde for 10 min and stained with 0.1 % crystal violet for 10 min. The number of colonies, defined as >50 cells/colony were counted.
Cell cycle assay
Forty-eight hours after transfection, MCF-7 and MDA-MB-231 cells were harvested in ice-cold phosphate-buffered saline (PBS) and then fixed with 70 % ice-cold ethanol. The fixed cells were incubated with 50 μg/ml propidium iodide (Sigma–Aldrich) and 50 μg/ml RNase A for 30 min at 37 °C in the dark and analyzed by FACScan (Becton Dickinson, USA).
Luciferase assays
For ectopic expression of CCND1, CCND3 ORFs with 3′-UTR were amplified by PCR and then cloned into pGL3 Vector (Promega) downstream of the Renilla luciferase cDNA. The primers selected were as follows: CCND1-3′UTR-wt-up: 5′-GCTGCGAAGTGGAAAC CATC-3′; CCND1-3′UTR-wt-dn: 5′-CCTCCTTCTGCACACATTTGAA-3′. Cells of 60–70 % confluence in 24-well plates were co-transfected with luciferase reporter vectors and miR-503 expressing vectors, and a 1 ng pRLSV40 Renilla luciferase construct was used for normalization. The relative dual-luciferase activity was assayed 48 h after transfection using the dual-luciferase assay kit according to the manufacturer’s protocol (Promega, Wisconsin, WI, USA).
Statistical analysis
All statistical analyses were analyzed with SPSS 17.0 (SPSS, Inc., Chicago, IL, USA) statistical evaluation for data analysis that was determined by Student’s t test and one-way ANOVA. Differences with P < 0.05 were considered statistically significant.
Result
MiR-503 expression was downregulated in breast cancer tissues and cell lines
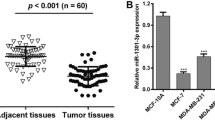
We compared the expression of miR-503 in breast cancer cells and patients samples, and we found that all six tested breast cancer cell lines showed significantly downregulated expression of miR-503 compared to the non-malignant breast epithelial cell MCF-3A, and the miR-503 expression was consistently downregulated in the breast cancer tissues compared with adjacent non-cancerous tissues (ANT) (Fig. 1). Taken together, our results suggested that miR-503 was underexpressed in breast cancer and might function as a tumor suppressor.

Expression of miR-503 in human breast cancer cell lines and clinical tissues. a Real-time PCR analysis of miR-503 expression in non-malignant breast epithelial cell MCF-3A and breast cancer cell lines, including MCF-7, T47D, MDA-MB-231, BT549, SKBR3, and ZR-75-30. b Relative miR-503 mRNA expression levels in eight paired human breast cancer tissues (T) and the adjacent non-cancerous tissues (ANT) from the same patient were detected by PCR analysis. Experiments were repeated at least three times (a and b). Each bar represents the mean of three independent experiments. *P < 0.05
MiR-503 inhibited cell proliferation of breast cancer
To further evaluate the effect of miR-503 on breast cancer growth, we transfected the MCF-7 and MDA-MB-231 cells with miR-503 mimics, miR-503-in, or the respective controls. Relative miR-503 expression was detected by using qRT-PCR and transfection of miR-503 restored its expression, while miR-503-in decreased its expression (Figs. 2a and 3a). In MTT and colon information assays, restoration of miR-503 in MCF-7 and MDA-MB-231 cells resulted in significant suppressing cell proliferation (Fig. 2b, c). In contrast, miR-503-in showed the opposite effect (Fig. 3b, c).

Overexpression of miR-503 inhibited breast cancer cell proliferation. a Validation of miR-503 expression levels after transfection by PCR analysis. b MTT assays revealed that upregulation of miR-503 inhibited growth of MCF-7 and MDA-MB-231 cells. c Quantification of crystal violet-stained cell colonies. d Flow cytometric analysis of the indicated MCF-7 and MDA-MB-231 cells transfected with miR-503 or miR-NC. Each bar represents the mean of three independent experiments. *P < 0.05

Inhibition of miR-503 promoted breast cancer cell proliferation. a Validation of miR-503 expression levels after transfection by PCR analysis. b MTT assays revealed that inhibition of miR-503 promoted growth of MCF-7 and MDA-MB-231 cells. c Quantification of crystal violet-stained cell colonies. d Flow cytometric analysis of the indicated MCF-7 and MDA-MB-231 cells transfected with miR-503 or miR-NC. Each bar represents the mean of three independent experiments. *P < 0.05
To further probe the regulatory mechanism of miR-503 inhibiting cell proliferation of breast cancer, we conducted a cell cycle assay. A higher proportion of MCF-7 and MDA-MB-231 cells transfected with miR-503 was in the G0/G1 phase and decreased in S phase compared with those transfected with relative controls, while miR-503-in showed the opposite effect. These results demonstrated that miR-503 arrests the cell cycle in the G0/G1 phase, thus inhibiting cell proliferation and then preventing further malignancy progression.
MiR-503 directly targeted CCND1 by binding to its 3′-UTR
Using TargetScan algorithms, we identified CCND1 as a candidate miR-503 target gene. To determine whether miR-503 could decrease CCND1 expression by targeting the predicted binding site, we inserted the CCND1 3′-UTR into pGL3 plasmid and a mutant of the putative binding site was also prepared (Fig. 4a). As showed in Fig. 4b, co-transfection of the miR-503 expression vector along with the CCND1 3′-UTR luciferase construct caused a significant reduction of luciferase activity as compared with the NC in MCF-7 and MDA-MB-231 cells. Meanwhile, miR-503 had no effect on the luciferase activity of CCND1 3′-UTR-mut type. Taken together, our results demonstrated that CCND1 was a direct target of miR-503 in breast cancer cells.

MiR-503 suppresses CCND1 expression by directly targeting the CCND1 3′-UTR. a Predicted miR-503 target sequence in the 3′-UTR of CCND1 (CCND1-3′-UTR) and positions of three mutated nucleotides (green) in the 3′-UTR of CCND1 (CCND1-3′-UTR-mut). b Luciferase reporter assay of MCF-7 and MDA-MB-231 cells transfected with the pGL3-CCND1-3′-UTR reporter or pGL3-CCND1-3′-UTR-mut reporter and miR-503. *P < 0.05. c Western blotting analysis of protein expression of CCND1, p21, p-pRb, and pRb in MCF-7 and MDA-MB-231 cells. β-Actin was used to serve as the loading control. d Levels of CCND1, p21, and p-pRb proteins were quantified using ImageJ software. β-actin was used as an internal control. The data are presented three independent experiments. *P < 0.05.
Given that our results indicated miR-503 could influence breast cancer cell proliferation, we examined its functions on the expression level of CCND1 downstream genes which regulate cell proliferation and cell cycle, including p21 and pRb. As showed in Fig. 4c, d, the expression level of the p21 protein was markedly upregulated, while p-pRb was decreased in miR-503 overexpressing cells compared with the negative control cells. In contrast, breast cancer cells transfected with miR-503-in showed the opposite effect. Altogether, our results demonstrated that CCND1 was a bona fide target of miR-503.
Discussion
In the current study, we demonstrated that miR-503 was decreased in breast cancer tissues and six cell lines. Furthermore, we found that ectopic overexpression of miR-503 inhibited the proliferation and cell cycle progression of breast cancer cells by arresting cells in the G0/G1 phase in vitro. Moreover, we demonstrated that miR-503 suppressed CCND1 expression via directly targeting its mRNA 3′-UTR. Taken together, our results suggest that overexpression of miR-503 might play an important role in inhibiting carcinogenesis and progression of breast cancer.
Increasing evidences indicated that microRNAs (miRNAs) are occurring, short, non-coding RNAs that control gene expression by repressing mRNA translation or by inducing mRNA degradation [21–23]. MiRNAs can contribute to tumor growth and progression by modulating the expression of many genes [24–26]. Previous studies have revealed that miR-503 expression is downregulated in several cancers, including osteosarcoma, lung cancer, hepatocarcinoma, and gastric cancer [16, 17, 19, 27]. However, it was uncertain whether dysregulation of miR-503 was associated with the progression of breast cancer. Here, our studies showed that expression of miR-503 was markedly downregulated in breast cancer cells and surgical breast cancer specimens. Gain- and loss-of-function studies indicated overexpression of miR-503 could suppress cell proliferation and colony formation and then induce cell cycle arrest breast cancer cells in the G0/G1 phase, suggesting miR-503 as a candidate tumor suppressor in the pathogenesis of breast cancer.
Using TargetScan algorithms, we identified CCND1 as a candidate miR-503 target gene. CCND1 is a crucial regulator of cell cycle that relates to the development of various cancers, which binds with cyclin-dependent kinases (CDK), which subsequently phosphorylates tumor suppressor protein Rb and then allows the cell cycle to progress through G1 into S phase, resulting in promoting cell proliferation [28–30]. In the present study, we found that CCND1 is a direct target of miR-503, which could downregulate its expression. Furthermore, we demonstrated that miR-503 inhibited breast cancer cell proliferation by binding to the 3′-untranslated region (UTR) of CCND1 mRNA. Further experiment showed that expression of p21 was upregulated and p-pRb were downregulated in MCF-7 and MDA-MB-231 cells transfected with miR-503. Altogether, our results indicated that miR-503 functionally modulates cellular proliferation and cell cycle regulators, p21 and pRb, thus relevant to cell proliferation.
Considered as a whole, the current study revealed that miR-503 operated as a potent tumor suppressor that downregulated the expression of CCND1, and then suppressing the cell proliferation of breast cancer, and this implies miR-503 to be a potential mediator for novel miRNA replacement therapy of breast cancer.
References
Siegel R, Desantis C, Jemal A. Colorectal cancer statistics. CA: Cancer J Clin. 2014;64:104–17.
Park YH, Lee SJ, Jung HA, Kim SM, Kim MJ, Kil WH, Lee JE, Nam SJ, Ahn JS, Im YH. Prevalence and clinical outcomes of young breast cancer (YBC) patients according to intrinsic breast cancer subtypes: single institutional experience in Korea. Breast (Edinburgh, Scotland) 2015.
Krop IE, Kim SB, Gonzalez-Martin A, LoRusso PM, Ferrero JM, Smitt M, et al. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:689–99.
Brandt J, Garne JP, Tengrup I, Manjer J. Age at diagnosis in relation to survival following breast cancer: a cohort study. World J Surg Oncol. 2015;13:429.
Migliaccio I, Malorni L, Hart CD, Guarducci C, Di Leo A. Endocrine therapy considerations in postmenopausal patients with hormone receptor positive, human epidermal growth factor receptor type 2 negative advanced breast cancers. BMC Med. 2015;13:280.
Chai J, Wang S, Han D, Dong W, Xie C, Guo H. MicroRNA-455 inhibits proliferation and invasion of colorectal cancer by targeting RAF proto-oncogene serine/threonine-protein kinase. Tumour Biol: J Int Soc Oncodevelopmental Biol Med. 2015;36:1313–21.
Feng S, Pan W, Jin Y, Zheng J. Mir-25 promotes ovarian cancer proliferation and motility by targeting LATS2. Tumour Biol: J Int Soc Oncodevelopmental Biol Med. 2014;35:12339–44.
Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–10.
Dalmay T. Mechanism of miRNA-mediated repression of mRNA translation. Essays Biochem. 2013;54:29–38.
van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–56.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66.
Shen S, Yue H, Li Y, Qin J, Li K, Liu Y, et al. Upregulation of miR-136 in human non-small cell lung cancer cells promotes Erk1/2 activation by targeting PPP2R2A. Tumour Biol: J Int Soc Oncodevelopmental Biol Med. 2014;35:631–40.
Hao Z, Yang J, Wang C, Li Y, Zhang Y, Dong X, et al. MicroRNA-7 inhibits metastasis and invasion through targeting focal adhesion kinase in cervical cancer. Int J Clin Exp Med. 2015;8:480–7.
Sun B, Yang M, Li M, Wang F. The microRNA-217 functions as a tumor suppressor and is frequently downregulated in human osteosarcoma. Biomed Pharmacother = Biomed Pharmacotherapie. 2015;71:58–63.
Sun Q, Zhao X, Liu X, Wang Y, Huang J, Jiang B, et al. Mir-146a functions as a tumor suppressor in prostate cancer by targeting Rac1. Prostate. 2014;74:1613–21.
Chong Y, Zhang J, Guo X, Li G, Zhang S, Li C, et al. MicroRNA-503 acts as a tumor suppressor in osteosarcoma by targeting L1CAM. PLoS One. 2014;9, e114585.
Wang T, Ge G, Ding Y, Zhou X, Huang Z, Zhu W, et al. MiR-503 regulates cisplatin resistance of human gastric cancer cell lines by targeting IGF1R and BCL2. Chin Med J. 2014;127:2357–62.
Polioudakis D, Abell NS, Iyer VR. miR-503 represses human cell proliferation and directly targets the oncogene DDHD2 by non-canonical target pairing. BMC Genomics. 2015;16:40.
Ruiz-Llorente L, Ardila-Gonzalez S, Fanjul LF, Martinez-Iglesias O, Aranda A. MicroRNAS 424 and 503 are mediators of the anti-proliferative and anti-invasive action of the thyroid hormone receptor beta. Oncotarget. 2014;5:2918–33.
Peng Y, Liu YM, Li LC, Wang LL, Wu XL. MicroRNA-503 inhibits gastric cancer cell growth and epithelial-to-mesenchymal transition. Oncol Lett. 2014;7:1233–8.
Valinezhad Orang A, Safaralizadeh R. Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. 2014;2014:970607.
Vidaurre S, Fitzpatrick C, Burzio VA, Briones M, Villota C, Villegas J, et al. Down-regulation of the antisense mitochondrial non-coding RNAs (ncRNAs) is a unique vulnerability of cancer cells and a potential target for cancer therapy. J Biol Chem. 2014;289:27182–98.
Li C, Xiong Q, Zhang J, Ge F, Bi LJ. Quantitative proteomic strategies for the identification of microRNA targets. Expert Rev Proteomics. 2012;9:549–59.
Humphries B, Yang C. The microRNA-200 family: small molecules with novel roles in cancer development, progression and therapy. Oncotarget 2015.
Liang H, Liu M, Yan X, Zhou Y, Wang W, Wang X, et al. miR-193a-3p functions as a tumor suppressor in lung cancer by down-regulating ERBB4. J Biol Chem. 2015;290:926–40.
Jiang J, Lv X, Fan L, Huang G, Zhan Y, Wang M, et al. MicroRNA-27b suppresses growth and invasion of NSCLC cells by targeting Sp1. Tumour Biol: J Int Soc Oncodevelopmental Biol Med. 2014;35:10019–23.
Yang Y, Liu L, Zhang Y, Guan H, Wu J, Zhu X, et al. MiR-503 targets PI3K p85 and IKK-beta and suppresses progression of non-small cell lung cancer. Int J Cancer J Int du Cancer. 2014;135:1531–42.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66.
Tashiro E, Tsuchiya A, Imoto M. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 2007;98:629–35.
Schaal C, Pillai S, Chellappan SP. The Rb-E2F transcriptional regulatory pathway in tumor angiogenesis and metastasis. Adv Cancer Res. 2014;121:147–82.
Acknowledgments
This work was supported by Science and Technology Planning Project of Guangdong Province (2012B031800055) and National Natural Science Foundation of China (31400858). All authors designed the study together, performed the experiment together, analyzed the data, and wrote the paper; all authors approved the final manuscript.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Long, J., Ou, C., Xia, H. et al. MiR-503 inhibited cell proliferation of human breast cancer cells by suppressing CCND1 expression. Tumor Biol. 36, 8697–8702 (2015). https://doi.org/10.1007/s13277-015-3623-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3623-8