
Abstract
Studies have shown that microRNAs (miRNAs) are involved in the malignant progression of human cancer. However, little is known about the potential role of miRNAs in breast carcinogenesis. miR-124 expression in breast cancer tissue was measured by quantitative real-time PCR (qRT-PCR). Target prediction algorithms and luciferase reporter gene assays were used to investigate the target of miR-124. Breast cancer cells growth was regulated by overexpression or knockdown miR-124. At the end of the study, tumor-bearing mice were tested to confirm the function of miR-124 in breast cancer. In this study, we demonstrated that the expression of miR-124 was significantly downregulated in breast cancer tissues compared with matched adjacent non-neoplastic tissues. We identified and confirmed that cyclin-dependent kinase 4 (CDK4) was a direct target of miR-124. Overexpression of miR-124 suppressed CDK4 protein expression and attenuated cell viability, proliferation, and cell cycle progression in MCF-7 and MDA-MB-435S breast cancer cells in vitro. Overexpression of CDK4 partially rescued the inhibitory effect of miR-124 in the breast cancer cells. Moreover, we found that miR-124 overexpression effectively repressed tumor growth in xenograft animal experiments. Our results demonstrate that miR-124 functions as a growth-suppressive miRNA and plays an important role in inhibiting tumorigenesis by targeting CDK4.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most frequently occurring malignancy and the second most frequent cause of cancer death in women in many countries [1]. Significant progress has been made toward increasing the overall breast cancer survival rate [2, 3]. However, improvements related to breast cancer metastasis have been discouraging, and only marginal improvements were identified in some reports [2]. Breast cancer is both genetically and histopathologically heterogeneous. Although many molecular triggers have been found to play a vital role in breast cancer development, the mechanisms underlying this process remain largely unknown. An understanding of the molecular mechanisms underlying the progression of breast cancer is crucial for developing effective treatments for this disease.
Recent studies have shown that microRNAs (miRNAs) are involved in the malignant progression of breast cancer [4–6]. miRNAs are a class of post-transcriptional regulators composed of short non-coding RNAs (∼22 nt) that bind to the complementary sequences in the 3′-untranslated region (3′-UTR) of multiple mRNA transcripts, which results in the silencing of target genes [7, 8]. Recent studies have shown that miRNAs act as tumor suppressors or oncogenes in cancer [9, 10]. Additional reports also indicate that miRNAs may have multiple functions in breast cancer metastasis. One of the most conserved miRNAs is miR-124, which is abundantly and specifically expressed in the nervous system. The expression of miR-124 is enriched in neurons but not astrocytes, and this expression level increases over time in the developing nervous system [11]. Recent reports have further demonstrated that deregulation of miR-124 is related to carcinogenesis. The expression level of miR-124 is significantly decreased in glioma, medulloblastoma, oral squamous cell carcinoma (OSCC) and hepatocellular carcinoma (HCC), and bladder cancer, which suggests a potential tumor suppressive function of miR-124 [12–17]. However, the function of miR-124 in breast cancer cell cycle, especially its role in human breast cancer patients, is not elucidated. Further, the molecular mechanisms utilized by miR-124 to modulate the malignant phenotype of breast cancer cells are not fully understood.
Cyclin-dependent kinase 4 (CDK4) is a master regulator of the cell cycle that belongs to the cyclin-dependent kinase family (CDK). These kinases govern cell cycle phase transitions in mammals and boost global gene transcription. CDK4 has been identified as the major oncogenic driver among cell cycle CDKs. Kinases are rendered hyperactive through a broad variety of mechanisms in human cancer, including frequent amplification or mutation of their genes [18–22]. Several tumor types, including leukemia, breast and lung cancers, are dependent on cyclin D-dependent kinase activity [23–26]. At the mechanistic level, recent studies have revealed pro-tumorigenic functions of CDK4/6 beyond cell cycle progression and relative miRNA can regulate CDK4 expression [17, 25–31]. However, to date, the function of CDK4 in breast cancer and the mechanism underlying its regulation remain poorly understood.
In this study, we found that miR-124 was one of the most frequently downregulated miRNAs in breast cancer tissues obtained from breast resections. miR-124 was able to significantly inhibit breast cancer cell proliferation and arrest the cell cycle by targeting CDK4 via its 3′-UTR region. Ectopic expression of CDK4 was able to partially reverse the inhibition of cell proliferation caused by miR-124. Furthermore, miR-124 expression was inversely correlated with the CDK4 protein level in breast cancer. We also found that miR-124 overexpression effectively repressed tumor growth in xenograft animal models. Thus, these data suggest that miR-124 may function as a tumor suppressor that targets CDK4 to influence the proliferation of breast cancer cells.
Materials and methods
Tissue specimens
Breast cancer and adjacent normal tissue samples were obtained with informed consent from patients who had undergone breast cancer surgery at the Affiliated Hospital of Nanjing Medical University, Changzhou No. 2 People’s Hospital, Changzhou, China, from 2006 to 2012. All the samples were shown to be correctly labeled clinically and pathologically and immediately frozen at −80 °C until use. This study was approved by the Research Ethics Committee of Nanjing Medical University.
Cell lines and cell culture
Human breast cancer cell lines (MCF-7, Bcap-37, and MDA-MB-435S) were obtained from the Central Lab of the Affiliated Hospital of Nanjing Medical University, Changzhou No. 2 People’s Hospital. The cell lines were cultured in RPMI Medium 1640 (GIBCO, Invitrogen) containing 10 % fetal bovine serum (FBS, Invitrogen) and were grown in a humidified 5 % CO2 incubator at 37 °C.
RNA extraction and expression analysis
Total RNA was extracted using TRIzol reagent (Invitrogen) and a mirVana miRNA Isolation Kit (Ambion) according to the manufacturer’s instructions. cDNA was synthesized with the Revert AidTM First Strand cDNA Synthesis Kit (Fermentas). Real-time PCR was performed using SYBR Green PCR Master Mix (Bio-Rad systems) on a Bio-Rad MyiQ Real-time RT-PCR system (Bio-Rad Systems, USA). For miR-124 detection, U6 snRNA was used as an internal control. For CDK4 mRNA detection, β-actin was used as a normalization control. The relative quantification value for each target gene was obtained using the comparative cycle threshold 2-ΔΔCt method. The primers used for PCR are listed in Supplementary Table S1.
Western blot
The cells were washed in phosphate-buffered saline (PBS), and proteins were extracted in RIPA buffer. Lysates were cleared by centrifugation, and protein concentrations were estimated using the Bio-Rad protein assay (Bio-Rad, Milan, Italy). Then, 50 μg of protein/lane was loaded onto an acrylamide gel and separated by SDS–PAGE under denaturing conditions. The separated proteins were then transferred electrophoretically (250 mA per blot, 80 min; Wet-trans-Blot SD, Bio-Rad) to a polyvinylidene fluoride (PVDF) membrane soaked in transfer buffer (25 mmol/L Tris, 192 mmol/L glycine; Sigma-Aldrich) and 20 % methanol v/v (Carlo Erba, Milan, Italy). Non-specific binding was blocked by incubation of the blots in 5 % non-fat dry milk (Bio-Rad) in TBS/0.1 % Tween (25 mmol/L Tris, 150 mmol/L NaCl, and 0.1 % Tween v/v; Sigma-Aldrich) for 60 min. After washing, the blots were incubated overnight at 4 °C with the primary antibody anti-CDK4 (CST), and anti-actin (CST) was used as a reference protein. After incubation with the primary antibodies and washing in TBS/0.1 % Tween, anti-mouse or anti-rabbit secondary antibody (both diluted 1:5000) was added (as appropriate) and incubated for 1 h at room temperature. Immunoreactive protein bands were detected by chemiluminescence using enhanced tetramethylbenzidine horseradish peroxidase color development solution (TMB). The films were then subjected to densitometric analysis using a Gel Doc 2000 system (Bio-Rad). Protein bands were detected with SuperSignal West Pico chemiluminescence substrate (Pierce) and processed with the GenTools software package. In each experiment, the same amount of protein was used, and each experiment was repeated independently at least three times.
Cell cycle assay
The cell cycle was analyzed by flow cytometry. Cells were harvested 48 h following transfection, washed with PBS, and fixed in 75 % ethanol at −20 °C. After overnight fixation, the cells were washed with PBS and stained with propidium iodide (Beckman Coulter, Fullerton, CA) for 30 min. Cell cycle analysis was performed using the BD Flow Cytometry System with FACSDiva software (BD Biosciences, Franklin Lakes, USA). The cell cycle distribution is presented as the percentage of cells in G1, S, and G2 phases. The data were analyzed with FlowJo v5.7.2.
Vector construction
To determine whether miR-124 regulates the expression of the human gene CDK4 by directly targeting its 3’UTR, the wild-type full length 3’-UTR of CDK4 containing the five putative miR-124 binding sites was amplified from the genomic DNA using primer pairs and subsequently cloned downstream of the Renilla luciferase gene in the pGL3 vector (Promega, Madison, USA) and designated as Vector-Luc-CDK4-3’UTR. Mutant vectors contained six sites deleted in the predicted binding sites—and were constructed by primer amplification and named as Vector-Luc-CDK4M-3’ UTR. The genomic segment including the mature miR-124 sequence was amplified and constructed into vector, named as vector-miR-124. The primers used for PCR are listed in table S1. The coding sequence of CDK4 was amplified from the cDNA and cloned into pcDNA3.1 (-) (Invitrogen, Carlsbad, USA).
Oligonucleotide transfection
Both miR-124 and the negative control for in vitro studies were synthesized by Genepharma (Shanghai, China). The sequences of the miR-124 mimics were 5’-UAAGGCACGCGGUGAAUGCC-3’ and 5’-CAUUCACCGCGUGCCUUAUU-3’, the sequence of the miR-124 inhibitor was 5’-GGCAUUCACCGCGUGCCUUA-3’, the sequence of negative control was 5’-UUCUCCGAACGUGUCACGUTT-3’, and the sequence of the miRNA inhibitor negative control was 5’-CAGUACUUUUGUGUAGUACAA-3’. The oligonucleotides were transfected using Lipofectamine 2000 reagent (Invitrogen) at a concentration of 80 nM.
Dual-Luciferase reporter gene assay
The luciferase reporter gene assay was performed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Cells that were 90 % confluent were seeded in 24-well plates. For the CDK4 3’UTR luciferase reporter assay, wild-type or mutant reporter constructs were co-transfected into MCF-7 cells in 24-well plates with 100 nM miR-124 and the Renilla plasmid using Lipofectamine 2000 (Invitrogen). Reporter gene assays were performed 48 h post-transfection using the Dual-Luciferase Assay System. Firefly luciferase activity was normalized to the corresponding Renilla luciferase activity to account for differences in transfection efficiency. All experiments were performed at least three times.
Stable transfection and in vivo animal studies
The agomiR-124 and antagomiR-124 expression constructs were generated by Genepharma (Shanghai, China). MCF-7/PBS cells, MCF-7/agomiR-124 cells, and MCF-7/antagomiR-124 cells were transfected with Lipofectamine 2000 reagent (Invitrogen), and miR-124 expression was confirmed by qRT-PCR. Four-week-old BALB/c nude mice were purchased from the Shanghai Experimental Animal Center (Chinese Academy of Sciences, Shanghai, China). A total of 30 mice were randomly divided into four groups, and each mouse was injected subcutaneously with 1 × 107 cells. The tumor volume (mm3) was measured every 7 days and was calculated using the following formula: volume = width × length × height/2. The animals were sacrificed 42 days after seeding the tumor cells. All tumor grafts were excised, weighed, and harvested. All animal experiments were performed with the approval of the Animal Care and Use Committee of Nanjing Medical University.
Statistical analysis
SPSS software (version 13.0, SPSS Inc., Chicago, IL, USA) was used for statistical analyses. Continuous data are presented as the mean ± SD and were compared between two groups using Student’s unpaired t test. The linear correlation coefficient (Pearson’s r) was calculated to determine the correlation between CDK4 and miR-124 expression in paired tissues. Kaplan-Meier analysis used to compare miR-124 expression for patient survival. p < 0.05 was considered to be statistically significant. All graphs were generated with GraphPad Prism Version 5.0.
Results
miR-124 was frequently downregulated in breast cancer and inhibited breast cancer cell proliferation by arresting the cell cycle
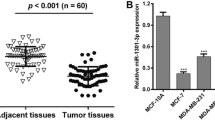
To explore the functional role of miR-124 in breast carcinogenesis, we first compared the expression levels in clinical breast carcinomas and paired adjacent normal tissues from 40 cases of breast cancer patients. By stem-loop quantitative real-time RT-PCR (qRT-PCR), we showed that the expression level of miR-124 was reduced in breast tumor specimens compared with adjacent normal tissues (Fig. 1a, p < 0.001 N = 40). This result indicated that reduced miR-124 expression was a frequent event in human breast cancer and may be involved in breast carcinoma progression.

miR-124 expression and function in breast cancer. a Relative expression of miR-124 in 20 breast cancer tissues compared with their pair-matched adjacent non-tumor tissues (normal). miR-124 was quantified by qRT-PCR with specific primers for miR-124 and U6 snRNA. b miR-124 expression in three breast cancer cell lines. Adjacent non-neoplastic tissues pooled from 20 samples were used as the control. A miR-124 mimic decreased proliferation in MCF-7 (c) and MDA-MB-435S (d) breast cancer cells compared with normal cells. A miR-124 inhibitor significantly increased the proliferation of MCF-7 (e) and MDA-MB-435S (f) breast cancer cells compared with normal cells. g DNA content was analyzed with propidium iodide (PI) staining and flow cytometry. miR-124 induced greater G1 cell cycle arrest of G0/G1 breast cancer cells compared with G0/G1 control cells. One representative histogram from three independent experiments with similar results is shown. The data are shown as the mean ± SD from three independent experiments. *p < 0.05
To determine the potential role of miR-124 in the progression of breast cancer, we detected miR-124 expression in the breast cancer cell lines MCF-7, MDA-MB-435S, and BCAP-37. miR-124 was expressed at significantly lower levels in these three cell lines compared with normal tissues pooled from 40 samples (Fig. 1b). Then, we transfected MCF-7 cells with a miR-124 mimic or inhibitor (Supplementary Figure S1). As expected, transfection of the miR-124 mimic decreased the proliferation of the MCF-7 and MDA-MB-435S breast cancer cell lines compared with normal control cells (transfected PBS as normal control) and cells transfected with miR-124 mimic control (Fig. 1c, d). In contrast, transfection of the miR-124 inhibitor significantly increased the proliferation of the breast cancer cell lines MCF-7 and MDA-MB-435S compared with normal control cells (transfected PBS as normal control) and cells transfected with miR-124 inhibitor control (Fig. 1e, f). These results demonstrated that ectopic expression of miR-124 inhibited cell proliferation in breast cancer cell lines. In addition to cell proliferation, we investigated the effect of miR-124 on cell cycle progression of breast cancer cells. The data showed that after transfection with the miR-124 mimic, the percentage of cells in the G0/G1 phase was increased from 34.8 to 41.2 % (Fig. 1g, p < 0.05). These results demonstrate that miR-124 inhibited breast cancer cell proliferation and arrested the cell cycle.
miR-124 represses CDK4 protein expression through the 3′-UTR
It is well known that miRNAs execute post-transcriptional regulation by binding to the 3’UTR of mRNAs. To elucidate the molecular mechanisms by which miR-124 inhibited the proliferation of breast cancer cells, miRNA targets were computationally predicted using two independent miRNA databases: TargetScan (http://www.targetscan.org/) and miRNAviewer (http://www.microrna.org/). Among the candidate target genes, we specifically focused on CDK4, which promotes cell cycle progression through the G1-phase into the S-phase. To examine the expression of CDK4 and its significance in breast cancer development, we measured its expression in breast carcinomas and paired adjacent normal tissues. The data showed that CDK4 expression was significantly higher in breast cancer tissues compared with paired adjacent normal tissues (Fig. 2a, p < 0.001 N = 40). These results indicated that increased CDK4 expression is a frequent event in human breast cancer that may be involved in breast carcinoma progression. CDK4 was also expressed at significantly higher levels in three breast cancer cell lines compared with normal tissues pooled from 40 samples (Fig. 2b). To validate that CDK4 is indeed directly targeted by miR-124, we investigated whether miR-124 recognizes the 3’UTR of CDK4 mRNA using a dual-luciferase reporter assay. Using the predictions from bioinformatics analysis (Fig. 2c), we cloned 3’UTR sequences containing the predicted wild-type (WT) target site or mutated target site (mutant) of CDK4 into the pGL3 control vector. We found that transfection of vector-miR-124 significantly suppressed the luciferase activity of the vector-CDK4 vector. By contrast, the repressive effect on the luciferase activity was ameliorated by mutations in the pGL3-CDK4 (vector-CDK4M) vector. These data confirm that CDK4 is a direct downstream target of miR-124 (Fig. 2d). These results indicated that the effect of miR-124 was due to specific and direct interaction with the putative binding sites on the 3’UTR of CDK4.

CDK4 was frequently upregulated in breast cancer and was a target of miR-124. a CDK4 expression was significantly higher in breast cancer tissues compared with the adjacent non-neoplastic tissues (normal). b CDK4 was expressed in the breast cancer cell lines MCF-7, MDA-MB-435S, and Bcap-37. Adjacent non-neoplastic tissues pooled from 20 samples were used as the control. c Bioinformatics predicted miR-124 target sequences within the 3’UTR of CDK4 mRNA. Several nucleotides were mutated within the seed region of the 3’UTR of CDK4. d Effect of miR-124 on CDK4 expression as determined by a luciferase reporter assay. The data were normalized by determining the ratio of firefly and Renilla luciferase activities measured at 24 h post-transfection. e, f MCF-7 cells were transfected with miR-124 mimic, inhibitor, mimic control, or inhibitor control (80 nM). The CDK4 protein level in MCF-7 cells was detected by Western blot. The bar graph represents the mean ± SD from three independent transfection experiments. *p < 0.05
Furthermore, we tested whether alteration of miR-124 levels affected CDK4 protein expression in breast cancer cells. As shown in Fig. 2e, overexpression of miR-124 inhibited CDK4 protein expression. In contrast, downregulation of miR-124 resulted in increased CDK4 protein levels (Fig. 2f). To test the effect of miR-124 on CDK4 mRNA, we measured the CDK4 mRNA level in breast cancer cells by qRT-PCR. No significant changes in CDK4 mRNA were found in MCF-7 cells transfected with miR-124 mimic or miR-124 inhibitor compared with the control (Supplementary Figure S2). Therefore, miR-124 directly targeted CDK4 mRNA and regulated CDK4 protein expression post-transcriptionally.
Downregulation of CDK4 expression inhibited breast cancer cell proliferation and arrested the cell cycle
To test the potential role of CDK4 in breast cancer progression, MCF-7 and MDA-MB-435S cells were cultured with SC-203873, a CDK4 inhibitor, for 3 days. Cell proliferation was significantly decreased by SC-203873 treatment compared with the control (Fig. 3 a, b). Further, the percentage of cells in the G1 phase was increased from 39.74 to 49.27 % after the MCF-7 cells were treated with the CDK4 inhibitor for 2 days (Fig. 3c). Taken together, these results suggested that the inhibition of CDK4 activity might attenuate the proliferation of breast cancer cells by blocking cell cycle progression.

Downregulation of CDK4 expression inhibited breast cancer cell proliferation and blocked cell cycle progression. Cell proliferation was significantly decreased by the CDK4 inhibitor in MCF-7 (a) and MDA-MB-435S cells (b) compared with the control. c DNA content was analyzed with propidium iodide (PI) staining and flow cytometry, and the data were used to investigate the cell cycle phase distribution. d Transfection of CDK4 vector/miR-124 mimic. The proliferation of breast cancer cells was partially rescued by transfection with a combination of CDK4 vector and miR-124 mimic compared with transfection of CDK4 expression vector alone or miR-124 mimic alone. e DNA content was analyzed with propidium iodide (PI) staining and flow cytometry, and the data were used to determine the cell cycle phase distribution. M indicates the mimic. The data are shown as the mean ± SD from three independent experiments. *p < 0.05
To explore whether miR-124 exerts its function through its target gene cdk4, we ectopically expressed CDK4 (using a CDK4 expression vector) together with a miR-124 mimic in breast cancer cells to determine whether there was compensation for or synergism with the effect of miR-124 on cell proliferation. Cell growth was significantly increased by CDK4 expression in MCF-7 cells compared with the control (Fig. 3d). When breast cancer cells were transfected with both CDK4 vector and the miR-124 mimic, their proliferation was partially inhibited compared with those transfected with the CDK4 expression vector alone (Fig. 3d). In addition to cell proliferation, the miR-124 mimic could also reverse the cell cycle progression induced by CDK4 in MCF-7 cells (Fig. 3e). The above data suggested that regulation of the growth of MCF-7 cells by miR-124 was mediated by targeting CDK4, which normally regulates cellular entry into the G1/S transition phase.
miR-124 levels were inversely correlated with CDK4 levels and relative to breast cancer patients overall survival
To confirm that miR-124 targets CDK4 in breast cancer, we examined the expression levels of miR-124 and CDK4 in total RNA extracted from 20 breast cancer tissues and paired adjacent normal tissues. We found that in both adjacent cancer tissues and in breast cancer tissues, the expression level of miR-124 was inversely correlated with the expression level of CDK4 (Supplementary Figure S3A and S3B, Fig. 4a). In addition, we also determined the expression level of miR-124 and CDK4 in the breast cancer cell lines MCF-7, BCAP-37, and MDA-MB-435S. The data revealed that miR-124 expression was lower than the expression of CDK4 in breast cancer cell lines (Fig. 4b). Furthermore, we found a significant inverse correlation when the expression level of CDK4 (tumor/adjacent) was plotted against the expression level of miR-124 (tumor/adjacent) in each patient (Fig. 4c). We also investigated the relationship between the expression level of miR-124 and prognosis in breast cancer patients. The median follow-up period for the patients studied was 67.1 months, with a range of 38 to 82 months, patients with high level of miR-124 expression (24 patients, 2 deaths) showed longer overall survival (median survival, 70.2 vs 47.2 months, p = 0.0368) than patients with low miR-124 expression (11 patients, 4 deaths). These data indicated that miR-124 downregulation might be associated with the increase in CDK4 levels and play an important role in breast cancer.

miR-124 levels were inversely correlated with CDK4 levels and relative to breast cancer patients overall survival. a Total RNA was extracted from tumor tissues or pair-matched adjacent non-tumor tissues (normal). The expression of miR-124 was inversely correlated with the expression of CDK4. b The expression of miR-124 was lower relative to the expression of CDK4 in breast cancer cell lines. c Correlation between the expression levels of miR-124 and CDK4 in different breast cancer cell types. A correlation coefficient (r) is shown
Stable overexpression of miR-124 inhibited tumor growth in vivo and was correlated with CDK4 expression
To determine the effect of miR-124 on tumor growth in vivo, we transfected MCF-7 cells with agomiR-124 (a modified miR-124 mimic used for in vivo studies), antagomiR-124 (a modified miR-124 inhibitor used for in vivo studies), or a negative control. Expression of miR-124 was observed in MCF-7 cells by Q-PCR (Fig. 5a). The three batches of cells were injected subcutaneously into nude mice. After 42 days, we observed slower tumor growth in the agomiR-124 group and faster tumor growth in the antagomiR-124 group compared with the control group (Fig. 5b). The average weight of tumors from the two groups was significantly different from that of the control group (Fig. 5c). To determine whether miR-124 affected breast cancer cell proliferation by targeting CDK4 in vivo, we investigated the CDK4 expression level in the tumor cells. CDK4 expression was significantly decreased in agomiR-124-transfected tumors, while CDK4 expression was significantly higher in the antagomiR-124 group compared with its levels in the control group (Fig. 5d). These results suggested that overexpression of miR-124 inhibited tumor growth by targeting CDK4 in vivo.

miR-124 inhibited xenograft tumor growth by targeting CDK4 in vivo. a The expression of agomiR-124 and antagomiR-124 was observed in MCF-7 cells by Q-PCR. b Groups of nude mice were implanted subcutaneously with MCF-7/PBS cells, MCF-7/agomiR-124 cells, or MCF-7/antagomiR-124 cells. Tumor volumes were recorded at the indicated times. c The tumor mass was determined when the mice were sacrificed. d CDK4 expression in tumors was examined by Western blot. The data are shown as the mean ± SD from three independent experiments. One representative tumor mass group from three independent experiments with similar results is shown. *p < 0.05
Discussion
Breast cancer is a type of malignant neoplasm originating from breast tissue. Worldwide, breast cancer is the most common cause of death among women. miRNAs have emerged as important regulators of post-transcriptional protein regulation, and global deregulation of miRNAs has been observed in various cancer tissues from numerous gene expression profile data sets [32, 33]. miRNAs can act as tumor suppressors if they target specific oncogenes, and the aberrant expression of tumor suppressive miRNAs may contribute to human carcinogenesis [34]. miR-124 was described as a brain-specific miRNA in mammals, and it may play a role in defining and maintaining neuron-specific characteristics [35, 36]. Recent studies have indicated that miR-124 targeted specific genes to regulate proliferation and migration in breast cancer [37–40].
Here, we show that the expression levels of miR-124 were significantly lower in human breast cancer tissues than in the adjacent non-neoplastic tissues. In functional studies, reintroduction of miR-124 dramatically repressed breast cancer cell proliferation, induced G1 cell cycle arrest of the G0/G1 cell population, and decreased the S and G2/M populations compared with the control. Additionally, a miR-124 inhibitor significantly increased breast cancer cell proliferation in vitro. Moreover, our study has identified CDK4 as the direct target of miR-124 in breast cancer cells. The expression level of CDK4 was significantly higher in human breast cancer tissues than in the adjacent non-neoplastic tissues.
These results are consistent with research on breast cancer, lung cancer, hepatocellular carcinoma, and cervical cancer that have identified CDK4 as a major oncogenic driver among members of the CDK superfamily. CDK4 becomes hyperactive in the majority of human cancers through a multitude of genomic alterations. Sustained activation of these protein kinases provides cancer cells with the ability to enter the cell cycle continuously by triggering G1-S-phase transitions and dramatically shortening the duration of the G1 phase. However, CDK4 also effectively counters cancer cell-intrinsic tumor suppression mechanisms, such as senescence and apoptosis, which must be overcome during cell transformation and remain inhibited throughout all stages of tumorigenesis. As a central “node” in cellular signaling networks, cyclin D-dependent kinases sense a plethora of mitogenic signals to orchestrate specific transcriptional programs [41]. In this study, we found that miR-124 could directly regulate the expression of CDK4 to modulate cell proliferation.
In conclusion, our observations suggest that a low level of miR-124 expression might result in elevated expression of CDK4. The increased CDK4 would allow breast cancer cells to proliferate in vitro and in vivo and would favor tumor progression. Additionally, our results showed that miR-124 inhibited breast cancer cell proliferation by regulating CDK4 expression and patients with high level of miR-124 expression showed longer overall survival. Our findings help to advance our understanding of the complex molecular mechanisms underlying the development and maintenance of miR-124/CDK4 levels that are associated with breast cancer. Based on these data, we suggest that the restoration of miR-124 activity may represent an attractive strategy for breast cancer therapy, and research should be focused on this area in the future.
References
Desantis C, Ma J, Bryan L, Jemal A: Breast cancer statistics, 2013. CA Cancer J Clin 2013.
Pagani O, Senkus E, Wood W, Colleoni M, Cufer T, Kyriakides S, et al. International guidelines for management of metastatic breast cancer: can metastatic breast cancer be cured? J Natl Cancer Inst. 2010;102:456–63.
van den Hurk CJ, Eckel R, van de Poll-Franse LV, Coebergh JW, Nortier JW, Holzel D, et al. Unfavourable pattern of metastases in m0 breast cancer patients during 1978-2008: a population-based analysis of the Munich Cancer Registry. Breast Cancer Res Treat. 2011;128:795–805.
Negrini M, Calin GA. Breast cancer metastasis: a microrna story. Breast Cancer Res. 2008;10:203.
Huang Q, Gumireddy K, Schrier M, le Sage C, Nagel R, Nair S, et al. The micrornas mir-373 and mir-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008;10:202–10.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microrna-10b in breast cancer. Nature. 2007;449:682–8.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. Microrna expression profiles classify human cancers. Nature. 2005;435:834–8.
Pillai RS. Microrna function: multiple mechanisms for a tiny rna? RNA. 2005;11:1753–61.
Slack FJ, Weidhaas JB. Microrna in cancer prognosis. N Engl J Med. 2008;359:2720–2.
Wang V, Wu W. Microrna-based therapeutics for cancer. BioDrugs. 2009;23:15–23.
Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS. A microrna array reveals extensive regulation of micrornas during brain development. RNA. 2003;9:1274–81.
Xia H, Cheung WK, Ng SS, Jiang X, Jiang S, Sze J, et al. Loss of brain-enriched mir-124 microrna enhances stem-like traits and invasiveness of glioma cells. J Biol Chem. 2012;287:9962–71.
Hunt S, Jones AV, Hinsley EE, Whawell SA, Lambert DW. Microrna-124 suppresses oral squamous cell carcinoma motility by targeting itgb1. FEBS Lett. 2011;585:187–92.
Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. Mir-124 and mir-203 are epigenetically silenced tumor-suppressive micrornas in hepatocellular carcinoma. Carcinogenesis. 2010;31:766–76.
Zheng F, Liao YJ, Cai MY, Liu YH, Liu TH, Chen SP, et al. The putative tumour suppressor microrna-124 modulates hepatocellular carcinoma cell aggressiveness by repressing rock2 and ezh2. Gut. 2012;61:278–89.
Deng X, Ma L, Wu M, Zhang G, Jin C, Guo Y, et al. Mir-124 radiosensitizes human glioma cells by targeting cdk4. J Neurooncol. 2013;114:263–74.
Zhang T, Wang J, Zhai X, Li H, Li C, Chang J. Mir-124 retards bladder cancer growth by directly targeting cdk4. Acta Biochim Biophys Sin (Shanghai). 2014;46:1072–9.
Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. Meyer zum Buschenfelde KH, Beach D: a p16ink4a-insensitive cdk4 mutant targeted by cytolytic t lymphocytes in a human melanoma. Science. 1995;269:1281–4.
Schmidt EE, Ichimura K, Reifenberger G, Collins VP. Cdkn2 (p16/mts1) gene deletion or cdk4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54:6321–4.
He J, Allen JR, Collins VP, Allalunis-Turner MJ, Godbout R, Day 3rd RS, et al. Cdk4 amplification is an alternative mechanism to p16 gene homozygous deletion in glioma cell lines. Cancer Res. 1994;54:5804–7.
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47.
Costello JF, Plass C, Arap W, Chapman VM, Held WA, Berger MS, et al. Cyclin-dependent kinase 6 (cdk6) amplification in human gliomas identified using two-dimensional separation of genomic DNA. Cancer Res. 1997;57:1250–4.
Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, et al. Requirement for cdk4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32.
Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin d1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22.
Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between k-ras oncogenes and cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73.
Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, et al. The requirement for cyclin d function in tumor maintenance. Cancer Cell. 2012;22:438–51.
Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, et al. A systematic screen for cdk4/6 substrates links foxm1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–34.
Zou X, Ray D, Aziyu A, Christov K, Boiko AD, Gudkov AV, et al. Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to arf/p53-independent senescence. Genes Dev. 2002;16:2923–34.
Ruas M, Gregory F, Jones R, Poolman R, Starborg M, Rowe J, et al. Cdk4 and cdk6 delay senescence by kinase-dependent and p16ink4a-independent mechanisms. Mol Cell Biol. 2007;27:4273–82.
Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–38.
Rane SG, Cosenza SC, Mettus RV, Reddy EP. Germ line transmission of the cdk4(r24c) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol. 2002;22:644–56.
Calin GA, Croce CM. Microrna signatures in human cancers. Nat Rev Cancer. 2006;6:857–66.
Iorio MV, Croce CM. Microrna dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143–59.
Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. Microrna-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–9.
Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The microrna mir-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mrna splicing. Mol Cell. 2007;27:435–48.
Cao X, Pfaff SL, Gage FH. A functional study of mir-124 in the developing neural tube. Genes Dev. 2007;21:531–6.
Han ZB, Yang Z, Chi Y, Zhang L, Wang Y, Ji Y, et al. Microrna-124 suppresses breast cancer cell growth and motility by targeting cd151. Cell Physiol Biochem: Int J Exp Cell Physiol, Biochem Pharmacol. 2013;31:823–32.
Li L, Luo J, Wang B, Wang D, Xie X, Yuan L, et al. Microrna-124 targets flotillin-1 to regulate proliferation and migration in breast cancer. Mol Cancer. 2013;12:163.
Li W, Zang W, Liu P, Wang Y, Du Y, Chen X, et al. Microrna-124 inhibits cellular proliferation and invasion by targeting ets-1 in breast cancer. Tumour Biol: J Int Soc Oncodevelopmental Biol Med. 2014;35:10897–904.
Liang YJ, Wang QY, Zhou CX, Yin QQ, He M, Yu XT, et al. Mir-124 targets slug to regulate epithelial-mesenchymal transition and metastasis of breast cancer. Carcinogenesis. 2013;34:713–22.
Huang Z, Choi BK, Mujoo K, Fan X, Fa M, Mukherjee S, Owiti N, Zhang N, An Z: The e3 ubiquitin ligase nedd4 negatively regulates her3/erbb3 level and signaling. Oncogene 2014.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81272323), the Natural Science Foundation of Jiangsu Province (BK2012590), the Technology Project of Changzhou Social Development (CE20125019, CE20125024, CE20135044), and the key project of the Changzhou Health Bureau (ZD201201, ZD201307).
Conflicts of interest
None
Author contributions
T.B. and C.Q. provided the original idea and were responsible for the study design, analysis and interpretation of data, statistical analyses, and writing of the manuscript; T.B., D.X., L.Y., N.X., K.Q., and Y.W. were responsible for the analysis and interpretation of data and statistical analysis; C.T., W.L., Y.N., and J.D. were responsible for technical, material support and statistical analyses; S.W. was responsible for data collection and technical supports; All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 283 kb)
Rights and permissions
About this article

Cite this article
Feng, T., Xu, D., Tu, C. et al. miR-124 inhibits cell proliferation in breast cancer through downregulation of CDK4. Tumor Biol. 36, 5987–5997 (2015). https://doi.org/10.1007/s13277-015-3275-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3275-8