
Abstract
Gliomas are the most malignant and aggressive primary brain tumor in adults. Despite concerted efforts to improve therapies, their prognosis remains very poor. Isocitrate dehydrogenase 1 (IDH1) mutations have been discovered frequently in glioma patients and are strongly correlated with improved survival. However, the effect of IDH1 mutations on the chemosensitivity of gliomas remains unclear. In this study, we generated clonal U87 and U251 glioma cell lines overexpressing the R132H mutant protein (IDH1-R132H). Compared with control cells and cells overexpressing IDH wild type (IDH1-WT), both types of IDH1-R132H cells were more sensitive to temozolomide (TMZ) and cis-diamminedichloroplatinum (CDDP) in a time- and dose-dependent manner. The IDH1-R132H-induced higher chemosensitivity was associated with nicotine adenine disphosphonucleotide (NADPH), glutathione (GSH) depletion, and reactive oxygen species (ROS) generation. Accordingly, this IDH1-R132H-induced growth inhibition was effectively abrogated by GSH in vitro and in vivo. Our study provides direct evidence that the improved survival in patients with IDH1-R132H tumors may partly result from the effects of the IDH1-R132H protein on chemosensitivity. The primary cellular events associated with improved survival are the GSH depletion and increased ROS generation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas are classified as grades I to IV on the basis of histopathological and clinical criteria established by the World Health Organization (WHO) [1]. Their specific histologic subtypes include astrocytomas, oligodendrogliomas, and ependymomas. High-grade gliomas are highly invasive tumors with high recurrent rate following treatment. Despite advances in surgical techniques, radiation therapy, and adjuvant chemotherapy, the prognosis of high-grade gliomas is still poor [2–4]. Therefore, in order to improve the efficacy and prognosis of treatment strategy, it is necessary to probe into the molecular mechanisms involved in occurrence and development of gliomas.
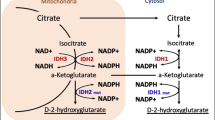
Isocitrate dehydrogenase 1 (IDH1) gene is located on chromosome 2 and encodes IDH1, which catalyzes the conversion of isocitrate to alpha-ketoglutarate (α-KG) and generates nicotine adenine disphosphonucleotide (NADPH) in the cytoplasm and peroxisomes [5]. IDH1 gene mutations occur frequently in patients with glioma. The most common mutation is R132H (>90 %) with R132C and R132S being less common [6–9]. Patients with IDH mutant tumors have a better prognosis than IDH wild-type (IDH1-WT) tumors, which has been confirmed in grades II–IV of gliomas [10–14]. For instance, Kaplan-Meier survival analysis revealed a highly significant association between IDH1 mutation and a better clinical outcome [13]. In patients with newly diagnosed glioblastoma multiforme (GBM) treated with radiation combined with temozolomide, Bujko et al. [15] estimated the prognostic value of the IDH1 mutations. They found that the 3-year overall survival was 60 % in the group with IDH1 mutation, while in the wild-type IDH1 group, it dropped to 29 % [15]. Among a large series of 382 patients, the anaplastic astrocytoma patients without IDH1 mutation did significantly worse than those with IDH1 mutation and even with glioblastoma patients with IDH1 mutation [16].
Since IDH1 mutations have been shown to be related to prolonged survival, there is an explosion of interest in the understanding of its contribution to response to chemotherapy in patients with GBM. Among 271 low-grade gliomas, a subgroup of 84 patients treated up-front with temozolomide (TMZ) was individualized [17]. IDH1 mutation was an independent predictor of an improved clinical outcome in response to treatment with TMZ [17]. These data indicate that IDH1 mutations may highlight the sensitivity to chemotherapy. However, when investigating the chemoresponse of IDH1 mutant glioma to procarbazine (Matulane), lomustine (CCNU), and vincristine (Oncovin), improved prognosis was found regardless of adjuvant therapy [18]. These findings support the hypothesis that IDH1 mutations lead to a better response to chemotherapy in glioma patients.
The IDH1 protein plays a significant role in cellular control of oxidative damage through the production of NADPH [19–21]. Sixty-five percent of the total NADPH production capacity in a glioblastoma patient is provided for by IDH activity. This is reduced by 38 % after the occurrence of R132 IDH1 mutation [10, 22]. Low-level cytoplasmic NADPH results in an impaired reduction of glutathione (GSH) and affects the thioredoxin system [23]. GSH is the most abundant intracellular antioxidant involved in the protection of cells against oxidative damage and in various detoxification mechanisms [24–26]. High-level of GSH in cells is related to apoptosis resistance [27]. On the contrary, depletion of intracellular GSH results in oxidative stress (generation of reactive oxygen species (ROS)), which is a known inducer of the transcription of specific genes involved in cell death [28]. ROS once generated will cause a massive oxidation of redox-sensitive proteins and lipids, leading to mitochondrial damage and inducing cell death through various signaling pathways [29]. However, the effect of IDH mutations on the above metabolic mechanism is limited.
In our previous study, we found that growth was significantly inhibited in glioma cells overexpressing the mutated IDH1 gene. Furthermore, these cells were characterized by increased intracellular NADPH levels accompanied by GSH depletion and ROS generation. Accordingly, the study demonstrated that using GSH-regulated mutant IDH1 glioma cells could obviously decrease the inhibition of cell growth. This study provides direct evidence that mutation of IDH1 profoundly inhibits the growth of glioma cells (unpublished). In the present study, therefore, we generated clonal U87 and U251 glioma cell lines overexpressing the R132H mutant protein (IDH1-R132H) to further evaluate the effect of IDH1 mutations on chemosensitivity of gliomas.
Materials and methods
Cell lines
Human glioblastoma cell lines U87 and U251 were purchased from Academy of Life Science, Shanghai, China, which were used for mutational analysis of the IDH1 genes. Cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (FBS; Invitrogen), 2 mm glutamine, and 100 units/ml penicillin/streptomycin in a 37 °C incubator with a humidified, 5 % CO2 atmosphere.
Site-directed mutagenesis and lentivirus production
IDH1 mutation constructs were generated with a site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). A wild-type IDH1 expression vector PGC-FU-IDH1-WT-FLAG was used as the mutagenesis template. IDH1 was mutated at Arg 132. Primer sequences used for mutagenesis are as follows: IDH1mu-Age I-F, GAGGATCCCCGGGTACCGGTCGCCACCATG TCCAAAAAAATCAGTGGCGG, and IDH1mu-Age I-R, TCACCATGGTGGCGACCG GAAGTTTGGCCTGAGCTAGTTTG. IDH1-WT and IDH1-R132H were cloned into PGC-FU-vector, which contains a biotin tag 5′ of the green fluorescent protein (GFP) tag. We obtained the PGC-FU-IDH1-WT-GFP and PGC-FU-IDH1-R132H-GFP, respectively. Lentiviral particles were added to U87 and U251 cells, and 48 h later, the infection efficiency was determined by analyzing GFP expression using flow cytometry (GeneChem).
Drug treatment
cis-Diamminedichloroplatinum (CDDP, MW 300), etoposide (VP-16, MW 589), and vincristine sulfate salt (VCR, MW 923) were purchased from Sigma. TMZ (MW 194) was provided by Schering-Plough Corporation, Canada. CDDP and VCR were dissolved in DW, VP-16, and TMZ in DMSO. Stock solutions were aliquoted and stored at −70 °C. Stock solutions were added to culture media to achieve the desired concentration. The cells were treated for a specified time without changing the media.
WST-1 cell viability assay
Cell lines were seeded in triplicate at a density of 3,200 cells per well in 96-well culture plates and incubated for 12 to 16 h at 37 °C and 5 % CO2. Chemotherapeutic agents, in various concentrations, were added to the cells and incubated for 24, 48, 72, 96, and 120 h. Control treatments consisted of equal volumes of DMEM. Following each culture time, 10 μl of WST-1 was added to each well, and the cells were incubated at 37 °C for an additional period of 1.5 h. Optical densities were determined on a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 450 nm. The percentage of cell viability was calculated as follows: Cell viability (%) = [A 450(sample) − A 450(blank)]/[A 450(control) − A 450(blank)] × 100.
Cellular NADPH and GSH levels
The cells were lysed by three repetitive freezing/thawing cycles to destroy the membrane of the cells for NADPH or GSH detection. NADPH was measured with Amplite™ Colorimetric NADP/NADPH Assay Kit (AAT Bioquest, Inc., USA) in a white/clear bottom 96-well plate using a NOVOStar microplate reader (BMG Labtech). The absorbance increase was monitored with an absorbance plate reader at 575 nm. Intracellular GSH was measured using the colorimetric microplate assay kits (Beyotime, Inc., Shanghai, China), according to the manufacturer’s instructions. Absorbance was read at 450 nm by the microplate reader.
Measurement of intracellular ROS
For analysis of intracellular ROS, the redox-sensitive fluorescent probe dichloro-dihydro-fluorescein diacetate (DCFH-DA) was used according to reactive oxygen species assay kit (Beyotime, Inc., Shanghai, China). Cells were incubated with 5 μM DCFH-DA for 30 min at 37 °C. The harvested cells were immediately analyzed at 485 nm excitation and 535 nm emission using a fluorescence spectrophotometer.
Tumorigenicity assay
Animal experiments were performed in accordance with the institutional animal care guidelines. Stably transfected U87-IDH1-R132H, U87-IDH1-WT, and control cells (1 × 105) were injected subcutaneously into the right front near the upper extremity of 16 BALB/C female nude mice (6 weeks old; the Experimental Animal Laboratories, Shanghai, China), while intravenous administration of TMZ or CDDP in the presence or absence of reduced glutathione sodium for injection (GSH). The resulting tumors were monitored weekly. The tumor volume (in cubic millimeters) was calculated using the standard formula: Length × Width × Height × 0.5236. All mice were sacrificed at the end of the 8th week after implantation. The tumors were harvested and individually weighed before fixation.
Statistical analyses
The results were expressed as mean ± standard error of the mean (SEM) and statistically compared with control group or within the groups using one-way ANOVA followed by Tukey’s multiple comparison test. Student’s t test was used to determine significance when only two groups were compared, and P < 0.05 was considered statistically significant.
Results
IDHI-R132H mutation significantly increases chemosensitivity to TMZ and CDDP
To evaluate the survival response mediated by IDH1-R132H mutation in astrocytoma cells, the control and the mutated cell lines were treated with chemotherapeutic agents (CTAs). Several CTAs (e.g., TMZ, VCR, VP-16, and CDDP) known to have clinical efficacy in the treatment of brain tumors were tested at clinically attainable concentration ranges. The U87-control, U87-IDH1-WT, and U87-IDH-R132H cells were exposed to varying concentrations of TMZ, CDDP, VCR, and VP-16, separately. The water-soluble tetrazolium salt (WST-1) cell viability test showed that the viability loss in IDH1-R132H cells increased dose and time dependently compared with the other two cell lines (P < 0.05, Fig. 1a, b) with the presence of TMZ or CDDP, but no significant difference was found after exposure to VCR or VP-16 (P > 0.05, Fig. 1c, d). To determine whether this effect could be observed in other isogenic glioma cell lines, we selected U251 cells, which have the similar sensitivity as U87 cells either to TMZ and CDDP or to VCR and VP-16 for the same experiments (Fig. S1).

U87-IDH1-R132H cells increased the sensitivity to TMZ and CDDP. Cells were treated with varying doses of TMZ (a), CDDP (b), VCR (c), and VP-16 (d) for different incubation periods. Viability was quantitated with WST-1 assay and expressed as the mean percentage of untreated control cells (mean ± SEM, n = 3). *P < 0.05; **P < 0.01, compared with control and IDH1-WT cells
IDH1 mutations reduce intracellular GSH and NADPH and increase intracellular ROS production in U87 Cells
With the U87-control, U87-IDH1-WT, and U87-IDH1-R132H cells, we investigated the metabolic mechanisms mediating the effect of IDH1-R132H protein overexpression on increasing sensitivity to TMZ and CDDP. Intracellular GSH plays important roles in maintaining redox status and defending oxidative stress, while NADPH is an essential cofactor for the regeneration of GSH by glutathione reductase. To determine whether the increased sensitivity to TMZ and CDDP was accompanied by decreased NADPH and GSH generation and increased ROS production, we investigated the levels of intracellular NADPH, GSH, and ROS in U87 cells transfected with PGC-FU-IDH1-R132H-GFP, PGC-FU-IDH1-WT-GFP, and PGC-FU-vector, separately. After the U87 cell lines were treated with 10 μg/ml TMZ and 10 μg/ml CDDP for 72 h, the levels of both NADPH and GSH decreased in IDH1-R132H cells compared to IDH1-WT and control cells (both P < 0.05, Fig. 2a, b). However, the level of intracellular ROS significantly increased in IDH1-R132H cells compared with the IDH1-WT and control cells (P < 0.05, Fig. 2c). The level of intracellular ROS decreased significantly after treatment with 1 mM GSH (P < 0.05, Fig. 2c).

A graphical representation of the U87-IDH1-R132H cells and the control cells. a The intracellular NADPH levels were measured as described in “Materials and methods.” *P < 0.05, significantly different from the control groups. b The intracellular GSH contents were determined according to the manufacturer’s instructions. The values are expressed in micromolar. *P < 0.05. c For analysis of intracellular ROS, cells were incubated with DCFH-DA, according to ROS assay kit. *P < 0.05
GSH protects the U87-IDH1-R132H cells from chemotherapy
To determine whether addition of GSH can protect the U87-IDH1-R132H cells from chemotherapy, we incubated the cells with GSH (1 mM) and TMZ (100 μg/ml) or CDDP (100 μg/ml) for different durations and determined cell viability by WST-1 assay. The increased sensitivities of U87-IDH1-R132H to TMZ and CDDP were both abrogated in the presence of 1 mM GSH, but the chemosensitivity of U87-IDH1-WT or U87-control cells was not affected by this dose of GSH (Fig. 3a, b).

GSH protected the U87-IDH1-R132H cells from chemotherapy. Cells were treated with GSH and TMZ (a) and GSH and CDDP (b) for different durations. Viability was determined by WST-1 assay and expressed as the mean percentage of untreated U87-control cells (mean SEM, n = 3). *P < 0.05; **P < 0.01, compared with U87-control and U87-IDH1-WT cells
IDHI-R132H mutation shows reduced tumorigenicity in nude mice following TMZ and CDDP treatment
IDH1-R132H overexpression in vitro shows significant chemosensitivity. We then asked whether IDH1-R132H overexpression in glioma cell lines following treatment with TMZ or CDDP affected their ability to initiate tumors in nude mice. The results show that compared with the U87-IDH1-WT and the control groups (Fig. 4a(a, b)), tumor size greatly decreases in mice inoculated with TMZ-treated U87-IDH1-R132H cells (Fig. 4(a–c)). As anticipated, the GSH-treated group develops tumors with a markedly larger size than the group inoculated with cells transfected with TMZ-treated U87-IDH1-R132H (Fig. 4(a–d)). There are significant differences in tumor weight and volume between them (Fig. 4b, c). Similar results were found for tumorigenicity following CDDP treatment in nude mice (data not shown).

In vivo inhibition of IDHI-R132H mutation in nude mice following TMZ and CDDP treatment. All mice were sacrificed at the 8th week after implantation, and tumors were harvested and individually weighed before fixation. a U87-IDH1-WT cells treated with TMZ. b U87-IDH1-control cells treated with TMZ. c U87-IDH1-R132H cells treated with TMZ. d U87-IDH1-R132H cells treated with TMZ and GSH. There were significant differences in tumor weight (b, c). Data are presented as mean ± SD. *P < 0.05
Discussion
IDH1 mutations were first reported in GBM in 2008 and later identified in a minority of patients with acute myeloid leukemia [30]. Such discovery has profound implications for the understanding and treatment of cancers. As a prevalent and specific biomarker, IDH1 mutations correlate with a better clinical outcome in the whole spectrum of gliomas and serve as an important diagnostic and prognostic factor. The patients with mutated IDH1 survived significantly longer [6]. IDH1 mutation may result in better prognosis for both low-grade gliomas (65 months for IDH1 vs. 38 months for wild type) and secondary GBMs (31 vs. 15 months) [16]. IDH1 mutations may function by affecting α-KG-dependent enzymes and be involved in epigenetic events. However, the mechanisms of mutant IDH1 in glioma genesis remain largely unknown. So far, the associations of IDH mutations with response to any type of treatment have been rarely validated [31].
In this study, we investigated whether IDH1 mutation can increase the sensitivity to CTAs in vitro and in vivo, as well as the possible mechanism of oxidative stress damage in glioma cell lines. The results showed that the IDH1-R132H mutation enhanced the cytotoxic effect of TMZ and CDDP in a dose- and time-dependent manner. No similar results were found after exposure to VP-16 and VCR. The reason may be related to the mechanism of CTAs. TMZ is a DNA-alkylating agent used for glioma chemotherapy, which relies on ROS production to work. TMZ-dependent ROS generation serves as an upstream signal and results in DNA damage, mitochondria damage, autophagy, or apoptosis [32, 33]. Low ROS production is related to chemoresistance to TMZ in glioma. Interference in the cellular redox balance with H2O2 and inhibitor of GSH can improve TMZ sensitivity and apoptosis [34]. CDDP is a heavy metal complex. It functions similar to alkylating agents. It acts on the purine and pyrimidine bases of DNA. CDDP can accumulate in mitochondria to improve the ROS production and activate JNK and MARK or ERK signaling pathway, subsequently resulting in cell death [35, 36]. Additionally, GSH/glutathione S-transferase (GST) is the main detoxification system of cells. CDDP is a substrate of GSTs, which can be pump out of the cell by GSH conjugate export pump (GS-X pump). GSH inhibitor can reverse CDDP resistance induced by GSH depletion [37]. However, there are no relevant reports that the mechanism-based chemotherapy for glioma of VP-16 and VCR is associated with GSH and ROS to date.
Our current findings are consistent with the report of improved survival in patients with IDH1-R132H mutant tumors, because TMZ and CDDP are the most common CTAs for glioma patients. Reportedly, IDH mutations are correlated with a higher rate of response to TMZ and predict longer survival in low-grade glioma patients [17]. However, the results from retrospective clinical studies regarding chemoresponse in IDH1 mutant glioma patients are conflicting. An isogenic U87MG model demonstrated that the overexpression of the IDH1-R132H mutant protein increased sensitivity to radiation, but not to TMZ [38], which is consistent with the study results of Dubbink et al. [11]. The reason for the conflicting results may be attributed to race and regional disparity, but our results may provide an important clue to further expose the treatment strategy for IDH1 mutant glioma.
To further investigate the metabolic mechanism of enhanced chemotherapy in the U87-IDH1-R132H cell lines, the levels of NADPH, GSH, and ROS in U87 cell lines were detected. The results show that mutant IDH1 may result in lower NADPH and GSH levels and higher ROS level, which formed a heightened oxidative response to TMZ and CDDP treatment in the U87-IDH1-R132H cells. NADPH, GSH depletion, and ROS generation may contribute to the intensified growth inhibition by TMZ and CDDP in the IDH1-R132H cells. Previous studies show that IDH1 mutations are associated with reduced catalytic activity of the IDH1 enzyme [39, 40], which hinders NADPH production. Moreover, mutated IDH1 consumes rather than produces NADPH, thus further lowering NADPH levels. The low NADPH levels could sensitize glioblastoma to irradiation and chemotherapy, thus explaining the prolonged survival of patients with mutated glioblastoma [10].
Oxidative stress is caused by an imbalance between the production of ROS and the ability of antioxidant defense system (AODS) to readily detoxify the reactive intermediates. The cells’ endogenous AODS scavenge ROS and thus maintain redox balance [26]. Most cancer cells are under oxidative stress associated with the increased metabolic activity and the production of high-level ROS. ROS generation may enhance the neoplastic behavior of a tumor by intensifying genetic instability and the capacity to invade host tissues [41]. Although increased ROS production plays an important role in maintaining cancer phenotype through stimulatory effects on cell growth and proliferation, it can also cause cellular damages including lipid peroxidation, DNA and protein oxidation, and enzyme inactivation [42]. The metabolically active cancer cells can produce high-level ROS and are under intrinsic oxidative stress, so they are more vulnerable to further oxidative stress caused by exogenous ROS-generating agents [43–46]. The GSH redox system is one key AODS involved in the protection of cells against oxidative damage and in various detoxification systems [47, 48]. Many therapeutic agents can enhance intracellular ROS generation [49–51], and the reduction in intracellular GSH is necessary for the formation of ROS [26, 52]. These results are consistent with our findings that glioma IDH1-R132H mutation enhances GSH depletion and ROS production in U87 and U251 cells in a dose- and time-dependent manner.
It is well documented that tumor cells have lower-level GSH [53]. Higher-level GSH has been associated with resistance to CTAs. In this study, we confirmed this finding with CTAs (e.g., TMZ and CDDP) and additional GSH in vitro and in vivo. The results showed that high-level GSH increased the resistance to CTAs. The increased sensitivities of U87-IDH1-R132H to both TMZ and CDDP were abrogated in the presence of GSH (Fig. 3). However, our results are based on the simulation experiments of glioma cells and thus need further clinical study to substantiate.
In conclusion, TMZ and CDDP showed a strong growth inhibitory effect toward U87 and U251 mutant cells. This selective toxicity of TMZ or CDDP between mutant and wild types may be due to a much higher level of oxidative stress in IDH1-R132H mutant-type glioma cells than the wild type. Additionally, targeted IDH1-R132H in glioma may be beneficial clinically and may enhance the efficacy of the currently used chemotherapeutic agents.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–49.
Su Y, Li G, Zhang X, Gu J, Zhang C, Tian Z, et al. JSI-124 inhibits glioblastoma multiforme cell proliferation through G(2)/M cell cycle arrest and apoptosis augment. Cancer Biol Ther. 2008;7(8):1243–9.
Yin D, Wakimoto N, Xing H, Lu D, Huynh T, Wang X, et al. Cucurbitacin B markedly inhibits growth and rapidly affects the cytoskeleton in glioblastoma multiforme. Int J Cancer. 2008;123(6):1364–75.
Jeon JY, An JH, Kim SU, Park HG, Lee MA. Migration of human neural stem cells toward an intracranial glioma. Exp Mol Med. 2008;40(1):84–91.
Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci U S A. 1999;96(18):10182–7.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12.
Sonoda Y, Kumabe T, Nakamura T, Saito R, Kanamori M, Yamashita Y, et al. Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci. 2009;100(10):1996–8.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73.
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol. 2009;27(34):5743–50.
Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, et al. The prognostic IDH1 (R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119(4):487–94.
Dubbink HJ, Taal W, van Marion R, Kros JM, van Heuvel I, Bromberg JE, et al. IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology. 2009;73(21):1792–5.
Metellus P, Coulibaly B, Colin C, de Paula AM, Vasiljevic A, Taieb D, et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol. 2010;120(6):719–29.
Yan W, Zhang W, You G, Bao Z, Wang Y, Liu Y, et al. Correlation of IDH1 mutation with clinicopathologic factors and prognosis in primary glioblastoma: a report of 118 patients from China. PLoS One. 2012;7(1):e30339.
Xie F, Tang JJ, Wang X, Liu YH, Mao Q. Correlation between IDH1 mutation and prognosis in supratentorial high-grade astrocytomas. Sichuan Da Xue Xue Bao Yi Xue Ban. 2013;44(2):184–7. 192.
Bujko M, Kober P, Matyja E, Nauman P, Dyttus-Cebulok K, Czeremszynska B. Prognostic value of IDH1 mutations identified with PCR-RFLP assay in glioblastoma patients. Mol Diagn Ther. 2010;14(3):163–9.
Hartmann C, Hentschel B, Tatagiba M, Schramm J, Schnell O, Seidel C, et al. Molecular markers in low-grade gliomas: predictive or prognostic? Clin Cancer Res. 2011;17(13):4588–99.
Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75(17):1560–6.
van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res. 2010;16(5):1597–604.
Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, et al. d-2-Hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012;26(18):2038–49.
Mohrenz IV, Antonietti P, Pusch S, Capper D, Balss J, Voigt S, et al. Isocitrate dehydrogenase 1 mutant R132H sensitizes glioma cells to BCNU-induced oxidative stress and cell death. Apoptosis. 2013;18:1416–25.
Kim J, Kim JI, Jang HS, Park JW, Park KM. Protective role of cytosolic NADP(+)-dependent isocitrate dehydrogenase, IDH1, in ischemic pre-conditioned kidney in mice. Free Radic Res. 2011;45(7):759–66.
Geisbrecht BV, Gould SJ. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem. 1999;274(43):30527–33.
Duran M, Kamerling JP, Bakker HD, van Gennip AH, Wadman SK. l-2-Hydroxyglutaric aciduria: an inborn error of metabolism? J Inherit Metab Dis. 1980;3(4):109–12.
Chuang JI, Chang TY, Liu HS. Glutathione depletion-induced apoptosis of Ha-ras-transformed NIH3T3 cells can be prevented by melatonin. Oncogene. 2003;22(9):1349–57.
Ghibelli L, Fanelli C, Rotilio G, Lafavia E, Coppola S, Colussi C, et al. Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J. 1998;12(6):479–86.
Guha P, Dey A, Sen R, Chatterjee M, Chattopadhyay S, Bandyopadhyay SK. Intracellular GSH depletion triggered mitochondrial Bax translocation to accomplish resveratrol-induced apoptosis in the U937 cell line. J Pharmacol Exp Ther. 2011;336(1):206–14.
Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58.
Kretz-Remy C, Arrigo AP. Gene expression and thiol redox state. Methods Enzymol. 2002;348:200–15.
Juan ME, Wenzel U, Daniel H, Planas JM. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J Agric Food Chem. 2008;56(12):4813–8.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–66.
Guo C, Pirozzi CJ, Lopez GY, Yan H. Isocitrate dehydrogenase mutations in gliomas: mechanisms, biomarkers and therapeutic target. Curr Opin Neurol. 2011;24(6):648–52.
Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH, Chen TH, et al. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PLoS One. 2012;7(6):e38706.
Kohsaka S, Takahashi K, Wang L, Tanino M, Kimura T, Nishihara H, et al. Inhibition of GSH synthesis potentiates temozolomide-induced bystander effect in glioblastoma. Cancer Lett. 2013;331(1):68–75.
Oliva CR, Moellering DR, Gillespie GY, Griquer CE, et al. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS One. 2011;6(9):e24665.
Schweyer S, Soruri A, Heintze A, Rzdzun HJ, Favvazi A. The role of reactive oxygen species in cisplatin-induced apoptosis in human malignant testicular germ cell lines. Int J Oncol. 2004;25(6):1671–6.
Yu ZY, Liang YG, Xiao H, Shan YJ, Dong B, Huang R. Melissoidesin G, a diterpenoid purified from Isodon melissoides, induces leukemic-cell apoptosis through induction of redox imbalance and exhibits synergy with other anticancer agents. Int J Cancer. 2007;121(9):2084–94.
Das GC, Bacsi A, Shrivastav M, Hazra TK, Boldogh I. Enhanced gamma-glutamylcysteine synthetase activity decreases drug-induced oxidative stress levels and cytotoxicity. Mol Carcinog. 2006;45(9):635–47.
Li S, Chou AP, Chen W, Chen R, Deng Y, Phillips HS, et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 2013;15(1):57–68.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–5.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44.
Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–8.
Hu Y, Rosen DG, Zhou Y, Feng L, Yang G, Liu J, et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem. 2005;280(47):39485–92.
Toyokuni S. Oxidative stress and cancer: the role of redox regulation. Biotherapy. 1998;11(2–3):147–54.
Kondo S, Toyokuni S, Iwasa Y, Tanaka T, Onodera H, Hiai H, et al. Persistent oxidative stress in human colorectal carcinoma, but not in adenoma. Free Radic Biol Med. 1999;27(3–4):401–10.
Devi GS, Prasad MH, Saraswathi I, Raghu D, Rao DN, Reddy PP. Free radicals antioxidant enzymes and lipid peroxidation in different types of leukemias. Clin Chim Acta. 2000;293(1–2):53–62.
Hileman EA, Achanta G, Huang P. Superoxide dismutase: an emerging target for cancer therapeutics. Expert Opin Ther Targets. 2001;5(6):697–710.
Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kappaB by directly targeting p65. J Biol Chem. 1998;273(50):33508–16.
Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov Today. 2010;15(15–16):668–78.
Sharma V, Joseph C, Ghosh S, Agarwal A, Mishra MK, Sen E. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol Cancer Ther. 2007;6(9):2544–53.
Pramanik KC, Boreddy SR, Srivastava SK. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS One. 2011;6(5):e20151.
Sun J, McKallip RJ. Plumbagin treatment leads to apoptosis in human K562 leukemia cells through increased ROS and elevated TRAIL receptor expression. Leuk Res. 2011;35(10):1402–8.
Franco R, Panayiotidis MI, Cidlowski JA. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J Biol Chem. 2007;282(42):30452–65.
Sun Y, St Clair DK, Xu Y, Crooks PA, St Clair WH. A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res. 2010;70(7):2880–90.
Acknowledgments
This study was supported by the Youth Fund of the National Natural Science Foundation of China (81201975; 81201979; 81201349), the Youth Fund of the Natural Science Foundation of Jiangsu Province (BK2012224), the Natural Science Foundation of China Ministry of Health (2010-2-025), the Natural Science Foundation of Jiangsu Department of Health (H201124), the Six Major Human Resources Project of Jiangsu Province (2011-WS-065; 2010-WS-038), and the Natural Science Foundation of Jiangsu Colleges and Universities Grant (11KJB320010).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding authors
Additional information
Animal experiments were performed in strict compliance with the institutional animal care guidelines.
Jinlong Shi and Baolan Sun contributed equally to the work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
U251-IDH1-R132H cells increased the sensitivity to TMZ and CDDP. Cells were treated with varying doses of TMZ (A), CDDP (B), VCR (C), and VP-16 (D) for different incubation periods. Viability was quantitated with WST-1 assay and expressed as mean percentage of untreated control cells (mean ± SEM, n = 3). *P < 0.05, **P < 0.01 compared with control, IDH1-WT cells. (JPEG 43 kb)
Rights and permissions
About this article

Cite this article
Shi, J., Sun, B., Shi, W. et al. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumor Biol. 36, 655–662 (2015). https://doi.org/10.1007/s13277-014-2644-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2644-z