
Abstract
Present study was conducted to elucidate the molecular regulation mechanisms and the critical genes involved in regulating wheat early responses to Cadmium (Cd) stress. Both ICP-AES and fluorescence labeling were used to find that the Cd2+ influx into wheat roots was significantly suppressed by pre-treatment with or in the presence of the Ca2+ channel blocker LaCl3, Verapamil and N-ethylmaleimide. RNA-seq technology was used to identify differentially expressed genes (DEGs) during 12 h of 100 μM Cd stress. Raw reads (n = 80,309,620 were obtained. 108,549 unigenes were identified and classified into 25 COG categories. 8584 DEGs were detected. Many DEGs were involved in defense and detoxification mechanisms including signaling protein kinases, transcription factors, metal transporters and biosynthesis-related enzymes. A Gene Ontology annotation analysis based on the DEGs indicated the presence of many categories including cellular process, cell part and binding, catalytic activity and transporter activity. The Kyoto encyclopedia of genes and genomes pathway analysis identified 107 terms that were enriched for all of the 1018 DEGs. Quantitative real-time PCR of 27 selected DEGs revealed that the expression patterns were consistent with the transcript abundance changes as identified by Solexa analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cadmium (Cd) is one of the most harmful and widespread heavy metals and is readily taken up by plants and accumulated in various tissues (Akhtera et al. 2014; Cao et al. 2014a, b; Chen et al. 2008). Moderate Cd uptake by plants could result in considerable Cd accumulation in the edible portion of crops, which would decrease the quality and yield of crops and pose a significant threat to human health (Hawrylak-Nowak et al. 2015; He et al. 2011; Li et al. 2012). Therefore, there is an urgent need to elucidate the mechanisms of Cd tolerance in plants and to develop crop varieties with high Cd tolerance (Chmielowska-Bak et al. 2013; Dong et al. 2007).
Cd is highly toxic to plants through the direct or indirect inhibition of biological processes, such as photosynthesis, transpiration, nutrient uptake, and gene expression regulation (Chan and Hale 2004). Plants poisoned by Cd often show symptoms of leaf rolling, chlorosis, retarded growth, early senescence and cell death (Fotjová and Kovařik 2000; Lomaglio et al. 2015; Zhang et al. 2015a, b; Zhao et al. 2006). To survive, plants develop specific Cd detoxification mechanisms. One of the primary Cd tolerance mechanisms by which plants reduce the level of Cd uptake is by constraining the Cd content to the roots (Akhtera et al. 2014; Gill and Tuteja 2011; Sun et al. 2013). Cd tolerance is a complex process involving many genes regulated by a variety of physiological pathways (Li et al. 2014; Parrotta et al. 2015).
Wheat is an important food crop, and it has been reported that wheat roots can accumulate large quantities of Cd, which could cause a potential health risk in polluted areas (Berkelaar and Hale 2000; Chan and hale 2004). Considerable effort has been invested into investigating Cd stress in wheat, particularly its accumulation, translocation, physiological and metabolic activity, and deposition in cells (Harris and Taylor 2013). However, little is known about the mechanism of wheat responses to Cd stress at the molecular level (Gao et al. 2015a, b). Previous studies have indicated that differences in plant stress responses are controlled by a range of gene regulatory mechanisms that may act in various response and defense systems (Wang et al. 2013). Generally, transcription factors, transport proteins and some other critical genes involved in certain signal transduction and secondary metabolite pathways are considered to be common stress-related transcripts that are activated in response to both biotic and abiotic stresses (Zhang et al. 2014a, b). However, there are some unique genes involved in the response to a specific stress (Urano et al. 2010). For example, in Arabidopsis thaliana under Cd treatment, it has been found that the major genes activated not only involve non-specific stress-induced responses but also specific pathways including sulfur assimilation (Maynaud et al. 2013; Cebeci et al. 2008).
The next-generation sequencing (NGS) technologies based on two primary platforms, Roche/454 and Solexa/Illumina was have been successfully used to analyze the molecular regulation mechanisms and acquire candidate genes involved in various stress conditions (Tao et al. 2012; Xu et al. 2015; Zhang et al. 2015a, b; Li et al. 2015; Bhati et al. 2015). But their application to investigate the comprehensive transcriptional changes in response to Cd stress in wheat root was limited. The present study is aimed to elucidate the molecular regulation mechanisms and the critical genes involved in regulating wheat early responses to Cd stress. An abundance of differentially expressed Cd responsive genes were quantified, and the enriched networks for regulating Cd stress were acquired in wheat. Additionally, expression profiling of some differentially regulated genes were validated by quantitative real time PCR (qRT-PCR). These results would facilitate further investigation of the mechanisms of Cd accumulation/tolerance in plants and open prospective for excavating novel genes and for the genetic improvement of plant tolerance to Cd stress.
Materials and methods
Plant materials and Cd treatments
Wheat variety Nannong 9918 was used in this study. Wheat seeds were incubated on moist gauze at 25 °C for germination. The germinated seeds were transferred into a 1/4 Hoagland nutrient solution, which was changed daily. When their roots were approximately 4 cm long, the seedlings were treated with 100 μM CdCl2 for 12 h. A parallel culture was grown without subcultivation at 25 °C as a positive control.
Pre-treatment with a metabolic inhibitor and ion channel blockers
To elucidate the transporter(s) responsible for mediating the Cd2+ influx, pharmacological experiments were carried out on wheat seedlings. Two pharmacological agents were chosen for this experiment. LaCl3 (a non-selective cation channel current (NSCC) blocker), Verapamil (a known Ca2+ channel blocker, Li et al. 2012) and N-ethylmaleimide (NEM, a -SH inhibitor) were used to modify the activity of selected plasma membrane transporters. All chemicals were purchased from Sigma.
These inhibitors were mixed with the 1/4 Hoagland nutrient solution to achieve their final concentrations that were as follows: Verapamil, 20 mM; LaCl3, 50 mM; NEM, 15 mM. All these concentrations were determined based on previous reports (Wang and Fisher 1999; Li et al. 2012). The control treatment was pre-exposed in pharmacological-free medium. The plants were pre-exposed in solutions containing the pharmacological agents for 24 h prior to the measurement of the Cd fluxes and the uptake experiment.
Fluorescence localization of Cd in the root apex
The Cd2+ probe Leadmium Green AM dye (Invitrogen, USA) was used to investigate the distribution of Cd in the roots of wheat seedlings pretreated with 100 µM Cd for 12 h. A stock solution of Leadmium Green AM was made by adding 50 μL of DMSO to one vial of the dye. This stock solution was then diluted to 1/10 with 0.85% NaCl. The roots were immersed in 20 mM Na2-EDTA for 15 min and then rinsed three times with deionized water. The washed roots were immersed in the stain solution for 2 h in the dark and then washed three times for 10 min for each time with 0.85% NaCl. Samples were observed using a confocal laser scanning microscope (ECLIPSE 90i, Japan) with excitation at 488 nm and emission at 500-550 nm, and serial confocal optical sections were taken.
Determination of Cd content
The samples were prepared based on the reference reported by Khan et al. (2013). After the wheat roots under 100 μM Cd stress for 48 h in the deletion or addition of 5 mM 3-MA, the root samples were harvested respectively. Then, the harvested samples were dried at 60 °C for 48 h and at 100 °C for 24 h in next step. After dying, the root samples were digested using the mixed HNO3/HCLO4 (5:1, v/v) solution. After mineralization, the Cd content in the roots was detected with the test of ICP-AES (IRIS/AP optical emission spectrometer, Thermo Jarrel Ash, San Jose, CA, USA).
cDNA library preparation and illumina sequencing
For RNA sequencing, 1 cm sections were taken from the tip of approximately 10–15 roots from 10 to 15 individual seedlings. The harvested tissues were immediately frozen in liquid nitrogen and stored at −80 °C.
Library construction and sequencing were performed according to the previously described method (Rogers et al. 2012). Total RNA was extracted from the root samples using the TRIzol reagent (Takara, Japan). Two wheat root cDNA libraries were constructed using an RNA-seq assay for paired-end transcriptome sequencing, which was performed by Beijing Biomarker Technologies (Beijing, China). Poly (A) mRNA was enriched from total RNA by using the NEBNext Poly (A) mRNA Magnetic Isolation Module (NEB, E7490). The NEBNext mRNA Library Prep Master Mix Set for Illumina (NEB, E6110 and NEBNext Multiplex Oligos for Illumina, NEB, E7500) was used for RNA-seq library construction, which was then was sequenced using Illumina HiSeq™ 2500.
Assessment of differential gene transcription
The raw reads generated by Illumina Hiseq™ 2500 were initially processed to obtain clean reads by removing the adapter sequences and low quality bases at the 3′ end. Simultaneously, the Q20, Q30 and GC-content of the clean data were calculated. All of the downstream analyses were based on high quality clean data. The resulting reads had a length of 40 to 100 bp (≥60% of all reads were 100 bp long) and quality scores of 26 or higher at all base positions. Read mapping was performed using the Wheat IWGSC survey sequence annotation (https://urgi.versailles.inra.fr/gb2/gbrowse/wheat_survey_sequence_annotation/). All high quality reads were mapped to the wheat reference genome [GCA_000818885.1]; the total assembly gap length was 3 kb using TopHat2 and Cufflinks (Trapnell et al. 2012). At least 64% of each read required a 90% similarity to the reference to be mapped; the read count for each gene was obtained from the mapping results.
The read counts were normalized to FPKM (reads per kilobase of exon model per million mapped reads) values (Trapnell et al. 2012) and log 10 transformed to meet the assumptions of the linear models. A differential expression analysis was performed using EBSeq (Rapaport et al. 2013). The P values were adjusted using the Benjamini–Hochberg method. The corrected P value of 0.05 was set as the threshold for significantly differential expression.
Functional annotation
To investigate genes differentially expressed and understand the critical genes in wheat roots responding to the Cd stress, clean reads of the 100 µM Cd and the control libraries were respectively mapped to the reference sequences and were assigned to unigenes and isoforms with the RSEM (RNA-seq by Expectation Maximization) software (Bhattacharyya et al. 2013).
The GO enrichment analysis of the DEGs was implemented using the GOseq package in R based on the Wallenius non-central hypergeometric distribution, which can adjust for gene length bias in differentially transcribed genes (DEGs). The KEGG pathway enrichment analysis of the DEGs was performed using KOBAS (KEGG Orthology-Based Annotation System). Functional classification of DEGs utilized the COG (Cluster of Orthologous Groups of proteins, http://www.ncbi.nlm.nih.gov/COG), GO (Gene Ontology, http://www.geneontology.org/) and KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.genome.jp/kegg/) databases.
The cut-off E-value was set at <10−15. For the NCBI non-redundant (nr) Swiss-Prot annotations, the BLAST2GO program was used to obtain the GO annotations of unique assembled transcripts for describing biological processes, molecular functions and cellular components; the Bonferroni-corrected P value (≤0.05) was used as the threshold for significance. GO terms fulfilling this condition were defined as significantly enriched GO terms in DEGs.
Cluster analysis
Transcription patterns were clustered using Cluster 3.0 with Euclidean distances and the hierarchical cluster method of complete linkage clustering and visualized with Java TreeView software.
Validation of DEG expression with quantitative reverse transcription PCR (qRT-PCR)
To validate the Illumina sequencing results, qRT-PCR analysis was performed. Samples and total RNAs were prepared using the previously described method. Twenty-seven genes were selected for qRT-PCR analysis with SYBR green-based real-time qRT-PCR using the ABI 7500 system (Applied Biosystems, Foster, CA, USA). Wheat tubulin was used as the endogenous control. cDNAs were analyzed in triplicate. Relative expression levels were calculated by the \(2^{{ - \Delta \Delta {\text{C}}_{\text{t}} }}\) method (Livak and Schmittgen 2001). Primers for the tested genes have been provided in the Supplementary Material Table S1.
Statistical analysis
Three independent experiments were performed. All data are presented as the mean ± standard deviation (SD). Variance analysis and comparison between two groups was analyzed using paired-samples t-tests using SPSS 11.0. Results were considered statistically significant when P < 0.05 (*).
Results and discussion
Localization of Cd in wheat root tips
The uptake of Cd into plant cells is known to be facilitated by Ca channels and low-molecular-weight SH-containing compounds (Perfus-Barbeoch et al. 2002). To test if the Ca channel and SH-binding ligands were involved in the Cd transport in wheat, the uptake of Cd was quantified in the presence of Ca-channel blocker (lanthanum as LaCl3 and Verapamil) or SH blocker (N-ethylmaleimide [NEM]). LaCl3 and Verapamil has been used as typical inhibitors of Ca channels and prevents Cd uptake in plants of tobacco, Arabidopsis, barley, etc. (Bourque et al. 2002; Horemans et al. 2007; Akhtera et al. 2014). And NEM, which can specifically block the proteins and small SH-containing compounds (e.g., glutathione), has been found to reduce Cd uptake in the root of the halophyte Suaeda salsa (Li et al. 2012).
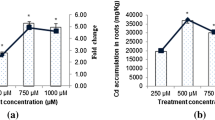
Furthermore, Leadmium Green AM has been successfully used to detect Cd in plant roots, it was employed to investigate the Cd distribution in wheat roots after 12 h of Cd exposure following pre-treatment with metabolic inhibitors and ion channel blockers. The fluorescent dye was loaded into the intact roots of wheat within 2 h and showed a clear, bright green fluorescence in the roots of Cd-treated plants (Fig. 1e), whereas almost no green fluorescence was observed in the control roots pre-treated with Cd for 0 h (Fig. 1a). A very low level of green fluorescence was observed in the roots in the absence of Cd (Fig. 1b), indicating that this dye did not react with divalent ions such as Ca2+ present in control roots. In addition, a greater intensity of fluorescence was observed near the root tips, indicating highly concentrated Cd, after exposure to 100 μM Cd for 12 h. In the roots pretreated with N-ethylmaleimide (NEM) (Fig. 1c), LaCl3 (Fig. 1d) and Verapamil (Fig. 1f), a reduced intensity of fluorescence was observed. These results suggested that the Cd influx into roots was suppressed by pre-treatment or by the presence of NEM, Verapamil and LaCl3, suggesting that Ca channels were involved in the uptake of Cd, whereas NEM inhibited Cd uptake only slightly. This conclusion is consistent with the observed decrease in Cd content of the wheat roots pre-treated with LaCl3, NEM and Verapamil following exposure to Cd for 12 h (Fig. 2).

Micrographs of roots from wheat root exposed to 100 μM Cd for 12 h. Roots from plants pre-treated with Cd for 0 h (a, b) and 12 h (e) were loaded with Leadamium Green AM dye for 2 h. Plants with pre-treatment of NEM (c), LaCl3 (d) and Verapamil (f) were then exposed to 100 μM Cd for 12 h before loading with Leadamium Green AM dye for 2 h. All images were taken at 20 magnification (Bar = 400 μm), and green fluorescence represents the binding of the dye to Cd

Cd accumulation in the wheat root after 12 h exposure in the solutions containing 100 μM Cd. Plants were pre-exposed to LaCl3, NEM and Verapamil, controls in Cd, Verapamil, LaCl3 and NEM free solutions. Values are mean ± S.D. (n = 3). Bars with different letters are significantly different (P < 0.05)
These results suggested the possible pathways of Cd uptake in wheat. The Cd influx into roots was significantly suppressed by pre-treatment with or by the presence of La3+ (a voltage-independent Ca channel blocker) and Verapamil (a specific calcium channel blocker). Both Verapamil and La3+ can inhibit Ca influx in plants by forming a very strong bond with the Ca channel (Li et al. 2012; Weiss 1974). And Cd accumulation in the wheat root had no significantly different between pre-exposed to LaCl3 and Verapamil. The results suggest that Cd uptake by wheat is likely regulated by Ca transporters or channels in the root cell plasm membrane. Because we observed the decreased Cd content in wheat roots pre-treated with the channel blocker followed by exposure to Cd for 12 h (Fig. 2). This finding provides evidence in support of the hypothesis that similar transport systems are involved in Cd uptake by wheat seedlings. NEM, as a thiol blocker, acts by binding with proteins and low-molecular-weight SH-containing compounds such as glutathione (Bobilya et al. 1992) and has a specific and irreversible interaction with thiol residues on proteins. NEM can thus inhibit Cd uptake in plants. Our result showed a similar inhibitory effect on Cd influx with previous reports when the roots was pretreated with NEM (Li et al. 2012).
Illumina sequencing analysis of wheat roots
Cadmium accumulation in plants is known to inhibit root growth. The wheat root as a whole is sensitive to Cd stress (Ci et al. 2010). Distinct patterns of transcription would be useful for exploring the molecular mechanisms of wheat root response to Cd stress. The Illumina HiSeq 2500 sequencing of the two wheat cDNA libraries, the untreated control (T01) and the 100 µM Cd stress treatment (T02), generated a large volume of data. The number of high-quality, clean paired-end sequencing reads for T01 and T02 was 40,306,993 and 40,002,627, respectively, with a total of 80,309,620 and 16,059,854,315 nucleotides acquired (Table 1) for the two pools. Among all the reads, 64–68% were readily mapped to positions in the wheat reference genome sequence (Chinese Spring). Due to the unavailability of complete wheat genome information, possibly resulting from high levels of repetitive sequences or insufficient read coverage, up to 26% of reads could not be mapped to the current wheat genome released by International Wheat Genome Sequencing Consortium (IWGSC) (Garbus et al. 2015). This issue potentially leads to missed reports of many stress-related genes. To minimize the influence of this gap in knowledge and map an informative, stress-related wheat transcriptome, we combined gene sequences collected from both public databases (including IWGSC, NCBI Unigene Database, and TriFLDB) and our de novo assembly; in total, 108549 non-redundant wheat unigenes were identified.
Exploration of differentially expressed genes (DEGs) in response to Cd stress
The assigned unigene and isoform expression levels were calculated using a normalizing statistic called FPKM (fragments mapped per kilobase of exon per million reads mapped), which provides a measure of expression level that accounts for variation in gene length (Bhattacharyya et al. 2013). A total of 8,584 DEGs were detected between the two libraries, and these DEGs included both upregulated (6963 transcripts) and downregulated genes (1621 transcripts) under the Cd treatment (Fig. 3). Among the 8,584 DEGs, most of them showed twofold to fivefold changes of their FPKM ratio (log 2 FC), while only a small portion (8.8%) of DEGs were greatly induced (more than fivefolds). The greatly induced DEGs were list out in Supplementary Table S2.

Volcano plot of gene expression differences between Cd100 (T02) and control samples (T01)
Gene Ontology (GO) and Pathway Functional Categorization of Cd Stress Responsive DEGs
All of the DEGs were analyzed with the GO function and pathway enrichment analysis using the GO classification system. Based on sequence homology, 7370 DEGs (85.86% of all DEGs) were assigned at least one GO term including 56 functional groups at the second level (Fig. 4). This result implies that a wide ranges of functional genes responsed to Cd stress.

Transcription patterns of Cd stress-regulated genes in the wheat root. T01 was the control sample with untreated wheat roots, and T02 was wheat roots treated with 100 μM Cd(Cl)2
At the first GO level, “cell part,” “cell,” “organelle,” “membrane” and “organelle part” terms were among the top five ranks in the cellular component category. For molecular function, “binding” and “catalytic activity” were the most abundant subcategories. While “cellular process” “metabolic process” and “response to stimulus” were the most highly represented in the biological process category. A variety of genes related to secondary products accumulation in “molecular function” and “biological process” were significantly enriched, including catalytic activity (GO:0003824), polyamine biosynthetic process (GO:0006596), carboxy-lyase activity (GO:0016831), aromatic-L-amino-acid decarboxylase activity (GO:0004058), tyrosine decarboxylase activity (GO:0004837), phenylacetaldehyde synthase activity (GO:1990055), chalcone isomerase activity (GO:0045430), phytoalexin biosynthetic process (GO:0052315), anthocyanin-containing compound biosynthetic process (GO:0009718), lignin biosynthetic process (GO:0009809), and positive regulation of flavonoid biosynthetic process (GO:0009963).
To find the most concentrated gene function groups in DEGs, the significantly enriched GO terms of DEGs annotation and the DEGs in significantly enriched GO terms were listed in Table 2. After cluster analyses, as shown in Fig. 5, T02 showed distinct transcription profiles of DEGs compared with T01 following Cd stress. The differential transcription trends of the DEGs in T01 and T02 may be related to the regulatory mechanism of Cd sensitivity in wheat roots.

Functional classification (GO) of Cd-regulated genes in wheat 9918
To determine whether the Cd stress-responsive genes were engaged in specific pathways, the DEGs were used as objects to search against the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. A total of 107 terms were enriched for all of the 1018 DEGs with pathway annotation. The top 20 significantly enriched pathways are shown in Fig. 6. Comparison of the Cd stress treatment with the control showed that 105 DEGs were enriched in ‘phenylpropanoid biosynthesis’, and 123 DEGs were related to ‘protein processing in endoplasmic reticulum’. This represents approximately 10.31 and 12.08% of the total genes that are involved in ‘phenylpropanoid biosynthesis’ and ‘protein processing in endoplasmic reticulum, respectively. The significantly enriched in KEGG pathways were listed in Table 3, and the DEGs which were significantly enriched in all the KEGG pathways are listed in Supplementary Table S3. The phenylpropanoid pathway serves as a rich source of metabolites in plants, being required for biosynthesis of lignin, flavonoids, coumarins and hydroxycinnamic acids. These secondary metabolites often play significant functions in plant defense. Aside from the structural function, lignin derivatives have been shown to have several bioactive functions. Ito et al. (2006) reported that lignin derivatives suppress the apoptosis of neural cells caused by oxidative stress. Flavoniods play a vital biological and pharmacological activities in in vitro studies (Cazarolli et al. 2008). The flavonoid biosynthetic pathways have already been reported in snapdragon (Antirrhinum majus), petunia (Petunia hybrida) A. thaliana, Z. mays and V. vinifera (Ma et al. 2015). Flavonoids are synthesized via the phenylpropanoid pathway, where the phenylalanine is used to produce 4-coumaroyl-CoA, and this then combined with malonyl-CoA to produce chalcones which are backbones of flavonoids, which can against pathogens attack. Sullivan et al. (2014) suggested that in the seed-to-seedling transition, phenylalanine (a key substrate in the phenylpropanoid pathway) may be a limiting factor in the development of initial mechanisms of UV protection in the developing leaf in soybean. These results provide valuable information for the future study of heavy metal stress response mechanisms in wheat.

The top 20 significantly enriched KEGG pathways of the annotated DEGs across T01 and T02. The left Y-axis indicates the strength of expression, X-axis indicates the enrichment factor. The right Y-axis indicates the KEGG pathway
All of the unigenes were also mapped to the COG database to further evaluate the effectiveness of the annotation process and understand gene function distribution characteristics of the species (Fig. 7). List of DEGs in all enriched COG pathways showed in Supplementary Table S4. These unigenes were classified into 25 COG categories. The ‘generation function prediction only’ category represented the most common category. Extracellular structures and nuclear structure represented the least common COG categories.

Cluster of Orthologous Groups of proteins (COG) function classification of unigenes in All-unigene. The horizontal coordinates are function classes of COG, and the vertical coordinates are the number of unigenes per class. The notation on the right shows the full name of the functions on the x-axis
Validation of illumina expression patterns by qRT-PCR analysis
To confirm the reliability of the Solexa analysis, 28 candidate DEGs representing a variety of expression patterns and biological functions (Table 4) were selected and their expression was detected using real-time quantitative PCR (qRT-PCR). The expression patterns from qRT-PCR showed general agreement with those from the Solexa sequencing (Table 4). The discrepancies in ratios are attributable to the different algorithms and sensitivities of the two techniques (Li et al. 2010; Shi et al. 2012). In the analysis of gene expression profiling, the deep-sequencing method generated absolute rather than relative expression measurements.
To further investigate and verify the variation in expression of the DEGs, transcriptional qRT-PCR analysis was performed on eight selected genes, including four upregulated (1-Traes_7DS_6A9DE4CB5, 2-Traes_7BL_EFF0E2E31, 3-Traes_1AS_36AF74187 and 4-Traes_6AL_FB41DAA2A) and four downregulated genes (5-Traes_5AL_6DC4E5956, 6-Traes_2BS_0D3F0D59A, 7-Traes_1AL_C1F546A56 and 8-Traes_1DL_7E5ED8683) exposed to a fixed concentration of Cd at 100 μM for different amounts of time (0, 4, 12, 24 and 36 h) for a more detailed temporal analysis. As shown in Fig. 8, the four downregulated DEGs were all downregulated at all levels of exposure to Cd stress. However, the expression of the four upregulated DEGs exhibited variation among the different exposure times: 2 showed upregulation after Cd stress for 4, 12 and 24 h, and then downregulation; 3 showed downregulation after 4 h and then upregulation after Cd stress for 12 and 24 h; and both 1 and 4 showed upregulation at each exposure time.

qRT-PCR analysis of eight selected DEGs with varying temporal exposure to a fixed concentration of 100 μM Cd
These qPCR results reflected significant alterations in major biological processes and metabolic pathways during Cd stress. This study represents the first comprehensive characterization of the molecular basis of the response to Cd stress in wheat, and provides useful information and a solid foundation for future investigations on the molecular regulation mechanism of Cd accumulation and tolerance in root vegetable crops.
DEGs related to signal sensing and transduction proteins
Whether Cd stress is perceived rapidly by the plant depends on Cd penetration into the plants through root uptake from soils or the aquatic environment. The root cell wall is directly in contact with metals in the soil solution (Mirzajani et al. 2013). When extra-cellular stimuli are encountered, the cell wall can activate a variety of specific stress-responsive signaling proteins to protect the cell from penetration at susceptible sites into the protoplast such as mitogen-activated protein kinases (MAPKs) and calcium-regulated protein kinases. In eukaryotes, MAPKs consist of three sequentially activated protein kinases including MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK, all of which are involved in responses to a variety of environmental, hormonal and developmental stimuli (Gawroński et al. 2014). In this study, 20 DEGs were identified that were highly homologous to genes encoding MAPKs such as MAPKK4, MAPKKK3, MAPK5, MPAK7, etc. With the exception of Traes_5DL_243735D6C, these DEGs were upregulated. Thirty-six DEGs were similar to calcium-binding protein genes including CML30, CML31, PBP1, CML25/26, etc., and most of these genes were upregulated under Cd stress. Activation of these genes may be advantageous during absorption of Cd ions by wheat roots.
DEGs related to transcription factors
It has been reported that transcription factors (TFs) play a central role in plant responses to abiotic stress by regulating downstream genes via specific binding to cis-acting elements in the promoters of target genes (Nakashima et al. 2012; Takasaki et al. 2010). Numerous TFs such as NAC, WRKY, basic leucine zipper (bZIP), ethylene-responsive factor (ERF) and myeloblastosis protein (MYB) have been documented to play a significant role in controlling the expression of specific stress-related genes (Thamilarasan et al. 2014; Chen et al. 2014). For example, 22 BrWRKY genes in Brassica rapa were differentially expressed in Chiifu compared to Kenshin under cold and drought stresses (Kayum et al. 2015); Overexpression of Arabidopsis SNAC-A genes such as RD26 and ATAF1, and rice SNAC-A genes such as OsNAC6 and OsNAC5 can improve drought and salinity tolerance; Overexpression of ThbZIP1 in tobacco can enhance the activity of both peroxidase (POD) and superoxide dismutase (SOD), and increase the content of soluble sugars and soluble proteins under salt stress conditions (Wang et al. 2010); overexpression of TaMYB19-B in wheat resulted in changes in several physiological indices and altered the expression levels of a number of abiotic stress-related genes, allowing the plants to overcome adverse conditions (Zhang et al. 2014a, b). Wheat TaERF1 was capable of binding to the GCC-box and CRT/DRE elements in vitro, Transcription of the TaERF1 gene was induced not only by drought, salinity and low-temperature stresses and exogenous ABA, ethylene and salicylic acid, but also by infection with Blumeria graminis f. sp. tritici. And overexpression of TaERF1 activated stress-related genes, including PR and COR/RD genes, under normal growth conditions, and improved pathogen and abiotic stress tolerance in transgenic plants (Xu et al. 2007). In the present study, a total of 298 DEGs, both up- and downregulated, that were identified as TFs, such as the WRKY family (i.e., WRKY41, 40, 33, 45, 33, 75, etc.), the ERF family (i.e., ERF118, 071, 109, 034, 017, 109, etc.), the MYB family (i.e., MYB 305, 4, 21, 2, 330, 39, etc.). These results suggested that both transcriptional activation and repression are involved and revealed that the differential expression trends of these TFs may contribute to the regulatory mechanism of Cd sensitivity in wheat roots.
DEGs related to metal transporters
Metal transporters could play a vital role in alleviating heavy metal toxicity by transporting metal ions out of the cell or sequestering them into the vacuole (Song et al. 2014). It has been reported that a wide range of transporter families including ATP binding cassette (ABC), natural resistance-associated macrophage proteins (Nramps), ZRT/IRT-like proteins (ZIPs) and the cation diffusion facilitators (CDFs) may contribute to heavy metal resistance (Bhati et al. 2015). In this study, 277 DGEs were identified as candidate genes involved as members of different metal transporter families, which were primarily related to ABC and peptide transporters. The results obtained here suggest that Cd uptake by wheat is regulated by Ca transporters or channels in root cell plasma membranes. This finding provides support for the hypothesis that similar transport systems are involved in Cd uptake by wheat. This conclusion is consistent with the observation of decreased Cd content in wheat roots pre-treated with the channel inhibitor LaCl3 prior to exposure to Cd for 12 h.
DEGs related to biosynthesis of chelating compounds and glutathione metabolism
The synthesis of metal-chelating compounds that can sequester and ultimately detoxify excess metal ions is another mechanism used by plants to combat heavy metal stress (Mendoza-Cózatl et al. 2005). Metallothioneins (MTs) are low-molecular-weight cysteine-rich metal-binding peptides, which are usually classified into four groups (MT1-4) (Yu et al. 2009). Recently, MT genes have been identified in a number of higher plants such as Arabidopsis and rice (Liu et al. 2015; Shahpiri et al. 2015). In the present study, 5 DEGs were homologs to genes encoding metallothionein-like protein 1. Phytochelatins (PCs) are another important class of heavy metal-binding ligands, which can bind metal ions via thiolate coordination. PCs are not formed as a direct result of the expression of a metal tolerance gene, but rather as the product of a biosynthetic pathway (Tan et al. 2015). Numerous physiological, biochemical and genetic studies have confirmed that glutathione (GSH) is the substrate for PC biosynthesis (Pomponi et al. 2006). The conversion of GSH to PCs can be catalyzed by a special c-glutamyl cysteine dipeptidyl transpeptidase (EC 2.3.2.15) called phytochelatin synthase (PCS). In the present study, 122 DEG sequences were found to encode for PCS, and 56 DEG sequences were found to encode for GSH. Based on the KEGG pathway assignment, 58 unigenes from the assembled de novo transcriptome were involved in glutathione metabolism. More than one unigene was annotated as the same enzyme, implying that such transcript sequences may represent different fragments of a single transcript or different members of a gene family (Esfahani and Shahpiri 2015). In most plant species, the Cd content of tissues tends to decrease in the following order: root, leaves, stem, inflorescence and seeds (Gao et al. 2015a, b; Zhao et al. 2010; Cao et al. 2014a, b). The results demonstrate that plants can effectively diminish Cd-induced damage by regulating their physiological and biochemical metabolism.
Conclusion
In the present study, Cd2+ influx into roots was significantly suppressed by pre-treatment with or in the presence of the Ca2+ channel blocker LaCl3 and the thiol blocker NEM, suggesting that Cd uptake by wheat roots is regulated by Ca transporters or channels in the root cell plasma membrane. Hundreds of DEGs expressed in response to Cd stress in wheat seedling roots. After gene annotation and blast, many DEGs were identified that were involved in defense and detoxification mechanisms including signaling protein kinases, transcription factors, metal transporters and biosynthesis-related enzymes, revealing their complex transcriptional regulation. Based on the differentially expressed genes, a Gene Ontology annotation analysis indicated the involvement of many gene categories including cellular process, cell part and binding, catalytic activity and transporter activity. The KEGG pathway analysis identified a total of 107 terms that were enriched for all of the 1018 DEGs such as ‘phenylpropanoid biosynthesis’ and ‘metabolic pathways’. The expression patterns of 27 selected genes involved in Cd tolerance derived from qPCR were consistent with their transcript abundance changes as identified by Solexa analysis. Identification of the potential DEGs involved in responses to Cd stress reflected significant alterations in major biological processes and metabolic pathways. Further functional analyses of these genes will promote our understanding of the molecular mechanisms underlying root adaptation to Cd stress.
Change history
02 May 2018
The authors are retracting this article [1] because Figs. 1A, 1E and 1F have been taken without permission from the Master’s thesis of Qiaoling Wang.
References
Akhtera MF, Omelon CR, Gordon RA, Moser D, Macfiea SM (2014) Localization and chemical speciation of cadmium in the roots of barley and lettuce. Environ Exp Bot 100:10–19
Berkelaar E, Hale B (2000) The relationship between root morphology and cadmium accumulation in seedlings of two durum wheat cultivars. Can J Bot 78:381–387
Bhati KK, Sharma S, Aggarwal S, Kaur M, Shukla V, Kaur J, Mantri S, Pandey AK (2015) Genome-wide identification and expression characterization of ABCC-MRP transporters in hexaploid wheat. Front Plant Sci 6:488
Bhattacharyya D, Sinha R, Hazra S, Datta R, Chattopadhyay S (2013) De novo transcriptome analysis using 454 pyrosequencing of the Himalayan Mayapple, Podophyllum hexandrum. BMC Genomics 14:748
Bobilya D, Briske-Anderson M, Reeve PG (1992) Zinc transport into endothelial cells is a facilitated process. J Cell Physiol 151:1–7
Bourque S, Lemoine R, Sequeira-Legrand A, Fayolle L, Delrot S, Pugin A (2002) The elicitor cryptogein blocks glucose transport in tobacco cells. Plant Physiol 130:2177–2187
Cao F, Chen F, Sun H, Zhang G, Chen ZH, Wu F (2014a) Genome-wide transcriptomeand functional analysis of two contrasting genotypes reveals key genes for cadmium tolerance in barley. BMC Genom 15:611
Cao F, Wang R, Cheng W, Zeng F, Ahmed IM, Hu X, Zhang G, Wu F (2014b) Genotypic and environmental variation in cadmium, chromium, lead and copper in rice and approaches for reducing the accumulation. Sci Total Environ 496:275–281
Cazarolli LH, Zanatta L, Alberton EH, Figueiredo MS, Folador P, Damazio RG, Pizzolatti MG, Silva FR (2008) Flavonoids: prospective drug candidates. Mini Rev Med Chem 8:1429–1440
Cebeci O, Kokturk B, Ergen N, Ozturkl L, Camak I, Budak H (2008) Differential expression of wheat transcriptomes in response to varying cadmium concentrations. Biol Plantarum 52:703–708
Chan DY, Hale BA (2004) Differential accumulation of Cd in durum wheat cultivars: uptake and retranslocation as sources of variation. J Exp Bot 55:2571–2579
Chen F, Wang F, Zhang GP, Wu FB (2008) Identification of barley varieties tolerant to cadmium toxicity. Biol Trace Elem Res 121:171–179
Chen N, Yang Q, Pan L, Chi X, Chen M, Hu D, Yang Z, Wang T, Wang M, Yu S (2014) Identification of 30 MYB transcription factor genes and analysis of their expression during abiotic stress in peanut (Arachis hypogaea L.). Gene 533:332–345
Chmielowska-Bak J, Lefevre I, Lutts S, Deckert J (2013) Short term signaling response in roots of young soybean seedlings exposed to cadmium stress. J Plant Physiol 15:1585–1594
Ci D, Jiang D, Wollenweber B, Dai T, Jing Q, Cao W (2010) Cadmium stress in wheat seedlings: growth, cadmium accumulation and photosynthesis. Acta Physiol Plant 32:365–373
Dong J, Mao WH, Zhang GP, Wu FB, Cai Y (2007) Root excretion and plant tolerance to cadmium toxicity – a review. Plant Soil Environ 53:193–200
Esfahani ES, Shahpiri A (2015) Thioredoxin h isoforms from rice are differentially reduced by NADPH/thioredoxin or GSH/glutaredoxin systems. Int J Biol Macromol 74:243–248
Fotjová M, Kovařik A (2000) Genotoxic effect of cadmium is associated with apoptotic changes in tobacco cells. Plant Cell Environ 23:531–537
Gao LS, Shun Z, Sheng NB, Yan X, Lai QL, Qing SC (2015a) The transportation and accumulation of arsenic, cadmium, and phosphorus in 12 wheat cultivars and their relationships with each other. J Hazard Mater 299:94–102
Gao W, Nan T, Tan G, Zhao H, Tan W, Meng F, Li Z, Li QX, Wang B (2015b) Cellular and subcellular immunohistochemical localization and quantification of cadmium ions in wheat (Triticum aestivum). PLoS ONE 10:e0123779
Garbus I, Romero JR, Valarik M, Vanžurová H, Karafiátová M, Cáccamo M, Doležel J, Tranquilli G, Helguera M, Echenique V (2015) Characterization of repetitive DNA landscape in wheat homeologous group 4 chromosomes. BMC Genomics 16:375
Gawroński P, Witoń D, Vashutina K, Bederska M, Betliński B, Rusaczonek A, Karpiński S (2014) Mitogen-activated protein kinase 4 is a salicylic acid-independent regulator of growth but not of photosynthesis in Arabidopsis. Mol Plant 7:1151–1166
Gill SS, Tuteja N (2011) Cadmium stress tolerance in crop plants e probing the role of sulfur. Plant Signal Behav 6:215–222
Harris NS, Taylor GJ (2013) Cadmium uptake and partitioning in durum wheat during grain filling. BMC Plant Biol 13:103
Hawrylak-Nowak B, Dresler S, Matraszek R (2015) Exogenous malic and acetic acids reduce cadmium phytotoxicity and enhance cadmium accumulation in roots of sunflower plants. Plant Physiol Bioch 94:225–234
He J, Qin J, Long L, Ma Y, Li H, Li K, Jiang X, Liu T, Polle A, Liang Z, Luo ZB (2011) Net cadmium flux and accumulation reveal tissue-specific oxidative stress and detoxification in Populus × canescens. Physiol Plantarum 143:50–63
Horemans N, Raeymaekers T, Beek KV, Nowocin A, Blust R, Broos K, Cuypers A, Vangronsveld J, Guisez Y (2007) Dehydroascorbate uptake is impaired in the early response of Arabidopsis plant cell cultures to cadmium. J Exp Bot 58:4307–4317
Ito Y, Shimazawa M, Akao Y, Nakajima Y, Seki N, Nozawa Y, Hara H (2006) Lig-8, a bioactive lignophenol derivative from bamboo lignin, protects against neuronal damage in vitro and in vivo. J Pharmacol Sci 102:196–204
Kayum MA, Jung HJ, Park JI, Ahmed NU, Saha G, Yang TJ, Nou IS (2015) Identification and expression analysis of WRKY family genes under biotic and abiotic stresses in Brassica rapa. Mol Genet Genomics 290:79–95
Khan MD, Mei L, Ali B, Chen Y, Cheng X, Zhu SJ (2013) Cadmium-induced upregulation of lipid peroxidation and reactive oxygen species caused physiological, biochemical, and ultrastructural changes in upland cotton seedlings. BioMed Res Int 2013:85–94
Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, Reidel EJ, Turgeon R, Liu P, Sun Q, Nelson T, Brutnell TP (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42:1060–1067
Li L, Liu X, Peijnenburg WJ, Zhao J, Chen X, Yu J, Wu H (2012) Pathways of cadmium fluxes in the root of the halophyte Suaeda salsa. Ecotoxicol Environ Saf 75:1–7
Li Y, Wang N, Zhao F, Song X, Yin Z, Huang R, Zhang C (2014) Changes in the transcriptomic profiles of maize roots in response to iron-deficiency stress. Plant Mol Biol 85:349–363
Li H, Yao W, Fu Y, Li S, Guo Q (2015) De novo assembly and discovery of genes that are involved in drought tolerance in Tibetan Sophora moorcroftiana. PLoS ONE 10:e111054
Liu J, Shi X, Qian M, Zheng L, Lian C, Xia Y, Shen Z (2015) Copper-induced hydrogen peroxide upregulation of a metallothionein gene, OsMT2c, from Oryza sativa L. confers copper tolerance in Arabidopsis thaliana. J Hazard Mater 294:99–108
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Lomaglio T, Rocco M, Trupiano D, De Zio E, Grosso A, Marra M, Delfine S, Chiatante D, Morabito D, Scippa GS (2015) Effect of short-term cadmium stress on Populus nigra L. detached leaves. J Plant Physiol 182:40–48
Ma J, Kanakala S, He Y, Zhang J, Zhong X (2015) Transcriptome sequence analysis of an ornamental plant, Ananas comosus var.bracteatus, revealed the potential unigenes involved in terpenoid and phenylpropanoid biosynthesis. PLoS ONE 10:e0119153
Maynaud G, Brunel B, Mornico D, Durot M, Severac D, Dubois E, Navarro E, Cleyet-Marel JC, Le Quéré A (2013) Genome-wide transcriptional responses of two metal-tolerant symbiotic Mesorhizobium isolates to zinc and cadmium exposure. BMC Genom 14:292
Mendoza-Cózatl D, Loza-Tavera H, Hernández-Navarro A, Moreno-Sánchez R (2005) Sulfur assimilation and glutathione metabolism under cadmium stress in yeast, protists and plants. FEMS Microbiol Rev 29:653–671
Mirzajani F, Askari H, Hamzelou S, Farzaneh M, Ghassempour A (2013) Effect of silver nanoparticles on Oryza sativa L. and its rhizosphere bacteria. Ecotoxicol Environ Saf 88:48–54
Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) NAC transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819:97–103
Parrotta L, Guerriero G, Sergeant K, Cai G, Hausman JF (2015) Target or barrier? The cell wall of early- and later-diverging plants vs cadmium toxicity: differences in the response mechanisms. Front Plant Sci 6:133
Perfus-Barbeoch L, Leonhardt N, Vavasseur A, Forestier C (2002) Heavy metal toxicity: cadmium permeates through calcium channels and disturbs the plant water status. Plant J 32:539–548
Pomponi M, Censi V, Di Girolamo V, De Paolis A, di Toppi LS, Aromolo R, Costantino P, Cardarelli M (2006) Overexpression of Arabidopsis phytochelatin synthase in tobacco plants enhances Cd2+ tolerance and accumulation but not translocation to the shoot. Planta 223:180–190
Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, Mason CE, Socci ND, Betel D (2013) Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol 14:R95
Rogers MF, Thomas J, Reddy AS, Ben-Hur A (2012) SpliceGrapher: detecting patterns of alternative splicing from RNA-seq data in the context of gene models and EST data. Genome Biol 13(1):R4
Shahpiri A, Soleimanifard I, Asadollahi MA (2015) Functional characterization of a type 3 metallolthionein isoform (OsMTI-3a) from rice. Int J Biol Macromol 73:154–159
Shi T, Gao Z, Wang L, Zhang Z, Zhuang W, Sun H, Zhong W (2012) Identification of differentially-expressed genes associated with pistil abortion in Japanese apricot by genome-wide transcriptional analysis. PLoS ONE 7:e47810
Song WY, Yamaki T, Yamaji N, Ko D, Jung KH, Fujii-Kashino M, An G, Martinoia E, Lee Y, Ma JF (2014) A rice ABC transporter, OsABCC1, reduces arsenic accumulation in the grain. Proc Natl Acad Sci USA 111:15699–15704
Sullivan JH, Muhammad D, Warpeha KM (2014) Phenylalanine is required to promote specific developmental responses and prevents cellular damage in response to ultraviolet light in soybean (Glycine max) during the seed-to-seedling transition. PLoS ONE 9:e112301
Sun J, Cui J, Luo C, Gao L, Chen Y, Shen Z (2013) Contribution of cell walls, nonprotein thiols, and organic acids to cadmium resistance in two cabbage varieties. Arch Environ Contam Toxicol 64:243–252
Takasaki H, Maruyama K, Kidokoro S, Ito Y, Fujita Y, Shinozaki K, Yamaquchi-Shinozaki K, Nakashima K (2010) The abiotic stress-responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol Genet Genomics 284:173–183
Tan SY, Jiang QY, Zhuo F, Liu H, Wang YT, Li SS, Ye ZH, Jing YX (2015) Effect of inoculation with glomus versiforme on cadmium accumulation, antioxidant activities and phytochelatins of Solanum photeinocarpum. PLoS ONE 10(7):e0132347
Tao X, Gu YH, Wang HY, Zheng W, Li X, Zhao CW, Zhang YZ (2012) Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam]. PLoS ONE 7:e36234
Thamilarasan SK, Park JI, Jung HJ, Nou IS (2014) Genome-wide analysis of the distribution of AP2/ERF transcription factors reveals duplication and CBFs genes elucidate their potential function in Brassica oleracea. BMC Genom 15:422
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7(3):562–578
Urano K, Kurihara Y, Seki M, Shinozaki K (2010) ‘Omics’ analyses of regulatory networks in plant abiotic stress responses. Curr Opin Plant Biol 13(2):132–138
Wang WX, Fisher NS (1999) Effects of calcium and metabolic inhibitors on trace element uptake in two marine bivalves. J Exp Mar Biol Ecol 236:149–164
Wang Y, Gao C, Liang Y, Wang C, Yang C, Liu G (2010) A novel bZIP gene from Tamarix hispida mediates physiological responses to salt stress in tobacco plants. J Plant Physiol 167(3):222–230
Wang Y, Xu L, Chen Y, Shen H, Gong Y, Limera C, Liu L (2013) Transcriptome profiling of radish (Raphanus sativus L.) root and identification of genes involved in response to lead (Pb) stress with next generation Sequencing. PLoS ONE 8:e66539
Weiss GB (1974) Cellular pharmacology of lanthanum. Annu Rev Pharmacol 14:343–354
Xu ZS, Xia LQ, Chen M, Cheng XG, Zhang RY, Li LC, Zhao YX, Lu Y, Ni ZY, Liu L, Qiu ZG, Ma YZ (2007) Isolation and molecular characterization of the Triticum aestivum L. ethylene-responsive factor 1 (TaERF1) that increases multiple stress tolerance. Plant Mol Biol 65(6):719–732
Xu Z, Liu C, Cai S, Zhang L, Xiong Z (2015) Heterologous expression and comparative characterization of vacuolar invertases from Cu-tolerant and non-tolerant populations of Elsholtzia haichowensis. Plant Cell Rep 34:1781
Yu J, Fujishiro H, Miyataka H, Oyama TM, Hasegawa T, Seko Y, Miura N, Himeno S (2009) Dichotomous effects of lead acetate on the expression of metallothionein in the liver and kidney of mice. Biol Pharm Bull 32:1037–1042
Zhang J, Feng J, Lu J, Yang Y, Zhang X, Wan D, Liu J (2014a) Transcriptome differences between two sister desert poplar species under salt stress. BMC Genom 15:337
Zhang L, Liu G, Zhao G, Xia C, Jia J, Liu X, Kong X (2014b) Characterization of a wheat R2R3-MYB transcription factor gene, TaMYB19, involved in enhanced abiotic stresses in Arabidopsis. Plant Cell Physiol 55:1802–1812
Zhang L, Pei Y, Wang H, Jin Z, Liu Z, Qiao Z, Fang H, Zhang Y (2015a) Hydrogen sulfide alleviates cadmium-induced cell death through restraining ROS accumulation in roots of Brassica rapa L. ssp. pekinensis. Oxid Med Cell Longev 2015 804603
Zhang M, Kong X, Xu X, Li C, Tian H, Ding Z (2015b) Comparative transcriptome profiling of the maize primary, crown and seminal root in response to salinity stress. PLoS ONE 10:e0121222
Zhao FJ, Jiang RF, Dunham SJ, McGrath SP (2006) Cadmium uptake, translocation and tolerance in the hyperaccumulator Arabidopsis halleri. New Phytol 172:646–654
Zhao FJ, Stroud JL, Eagling T, Dunham SJ, McGrath SP, Shewry PR (2010) Accumulation, distribution, and speciation of arsenic in wheat grain. Environ Sci Technol 44(14):5464–5468
Acknowledgements
We thank the other members of our laboratory for help in the research and for insightful remarks. This work was supported by the National Science Foundation of China (No. 31501234) and the Program of Yantai Entry-Exit Inspection and Quarantine Bureau (SK201419). Thank the referees for helpful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jieyu Yue, Xin Zhang and Ning Liu declare that they have no conflict of interest.
Studies with human or animal research
This article does not contain any studies with human subjects or animals performed by any of the authors.
Additional information
The authors are retracting this article because Figures 1A, 1E and 1F have been taken without permission from the Master’s thesis of Qiaoling Wang, “The mechanism of Cd stress on the physiological and cytotoxicity of onion seedlings” submitted to the College of Life Sciences, Tianjin Normal University in April 2014. All authors agree to this retraction.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Yue, J., Zhang, X. & Liu, N. RETRACTED ARTICLE: Cadmium permeates through calcium channels and activates transcriptomic complexity in wheat roots in response to cadmium stress. Genes Genom 39, 183–196 (2017). https://doi.org/10.1007/s13258-016-0488-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-016-0488-1