
Abstract
A methylation-sensitive amplification polymorphism method based on capillary electrophoresis was used to analyze DNA methylation levels in three cotton accessions, two salt-tolerant accessions CCRI 35 and Zhong 07 and one salt-sensitive accession CCRI 12. Many categories of DNA methylation happened in the three cotton accessions under salt treatment, including hypermethylation, hypomethylation, and other patterns. Hypermethylation happened at a significantly higher rate than that of hypomethylation in salt-tolerant accessions CCRI 35 and Zhong 07. On the contrary, in salt-sensitive accession CCRI 12, hypomethylation happened at a significantly higher rate than that of hypermethylation. In general, the global DNA methylation level significantly increased under salt stress in both salt-tolerant accessions CCRI 35 and Zhong 07, whereas there was no significant difference in the salt-sensitive accessions CCRI 12. Our results suggested that salt-tolerant cotton might have a mechanism of increasing the methylation level when responding to salt stress; the increase of the global level of DNA methylation and also different methylation patterns might play important roles in tolerance to salt stress in cotton. Some interesting genes were found through cloning and analysis of differently methylated DNA sequences, which might contribute to salt tolerance in cotton.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soil salinity is a major abiotic stress in plant agriculture worldwide that has led to research into salt tolerance with the aim of improving crop plants (Zhu 2001). It is estimated that approximately 20 % of all agricultural land and 50 % specifically of cropland in the world is under salt stress (Flowers and Yeo 1995). Besides natural causes such as salty water near around the coasts and contamination from the parental rocks and oceanic salts, cultivation practices have exacerbated increasing concentration of salts in the rhizosphere (Mahajan and Tuteja 2005). Therefore, salinization of arable land is getting more widespread and thus decreasing the yield of formerly productive soils throughout the world. The efforts to increase salt tolerance of crop plants bear extraordinary importance to maintain or improve the yield of agriculture on marginal lands and could potentially improve crop yield overall. The plant response to salinity consists of numerous processes that must function in coordination to alleviate both cellular hyperosmolarity and ion disequilibrium. Acclimation of plants to salinized conditions depends upon activation of cascades of molecular networks involved in stress sensing, signal transduction, and the expression of specific stress-related genes and metabolites. With the growing body of information about molecular markers, genomics, and post-genomics, significant breakthroughs have emerged in determining the mechanism and control of salinity tolerance at the molecular level (Türkana and Demiral 2009).
DNA methylation, an important epigenetic phenomenon, plays a vital role in tuning gene expression during plant development as well as in response to environmental stimuli (Lukens and Zhan 2007), allowing relatively rapid adaptation to new conditions without changing the DNA sequence in plants (Tan 2010). DNA methylation affecting the regulation of plant gene expression in response to environmental stresses is widely studied, including stresses such as cold (Shan et al. 2013), drought (Tan 2010; Wang et al. 2011a), salt (Dyachenko et al. 2006; Li et al. 2009; Tan 2010; Wang et al. 2011b; Zhao et al. 2010; Mastan et al. 2012; Bilichak et al. 2012), and metals (Aina et al. 2004; Kimatu et al. 2011). Although heritable methylation patterns and phenotypic variations that arise in response to stress are of potential value for plant breeding, their exploitation presents great challenges.
Many kinds of techniques have been adopted to analyze DNA methylation which generally can be assorted into two groups: detection of DNA methylation in whole genomes and specific site. Regarding the whole-genome detection, two isoschizomers that differ in their sensitivity to methylation of their recognition sequences HpaII and MspI are frequently used to detect cytosine methylation. Both enzymes recognize the tetranucleotide sequence 5′-CCGG. However, HpaII is inactive if one or both cytosines are fully methylated (both strands methylated) but cleaves the hemimethylated sequence (only one DNA strand methylated), whereas MspI cleaves C5mCGG but not 5mCCGG (McClelland et al. 1994). Reyna-López et al. (1997) modified the amplified fragment length polymorphism (AFLP, Vos et al. 1995) technique using HpaII and MspI instead of MseI in order to determine DNA methylation in fungi. Xiong et al. (1999) developed the method of methylation-sensitive amplified polymorphism (MSAP), adapted from the technique of Reyna-López et al. (1997), and investigated the cytosine methylation status of the rice genome. Xu et al. (2000) further optimized the adapters and primers in the method of MSAP which increased the efficiency of polymerase chain reactions (PCR) amplification and improved the resolution of AFLP banding pattern and the intensity of bands. With the introduction of fluorescence-labeled primers, researchers also proposed fluorescent-labeled methylation-sensitive amplified polymorphism (Xu et al. 2005).
Capillary electrophoresis (CE) is electrophoresis performed in a capillary tube. It is a very efficient separation technique that is available for the analysis of both large and small molecules. The transformation of conventional electrophoresis to modern CE was spurred by the production of inexpensive narrow-bore capillaries for gas chromatography and the development of highly sensitive on-line detection methods for high-performance liquid chromatography. Consequently, CE has the characteristics of high sensitivity, high resolution, and high speed compared to conventional electrophoresis.
As a very important source of fiber, cotton has a comparatively high level of salt tolerance with good economic returns which makes it a pioneer crop suitable to be planted in saline–alkali soil if its salt tolerance can be further improved. It is reported that the soil salinity content below 0.2 % will benefit growth of cotton, whereas cotton growth will be hindered when soil salinity content increases up to 0.3 % (Jiang et al. 2006). However, more work needs to be done so as to clearly elucidate the molecular mechanism of salt tolerance from different viewpoint in cotton. In this research, DNA methylation of different cotton accessions under salt stress was detected with MSAP based on CE method, so as to reveal epigenetic control of the molecular mechanism (s) of salt tolerance in cotton.
Materials and methods
Cotton materials and treatment conditions
The cotton accessions were obtained from the National Cotton Medium-Term Germplasm Bank, Cotton Research Institute, Chinese Academy of Agricultural Sciences (CCRI). Three cotton accessions, namely two salt-tolerant accessions CCRI 35 and Zhong 07, and one salt-sensitive accession CCRI 12 (Zhang et al. 2010) were used in this experiment.
For MSAP analysis, twenty seeds of each accession were selected and soaked in water for 24 h. The seeds were transferred to a layer of filter paper in a petri dish, covered with another wet filter paper, and then pure water was sprayed on the filter paper once a day to hasten germination. The germinated seeds were then planted in plastic pots containing washed sand under conditions of 14-h-light/10-h-dark with the air temperature of 30 °C-light/25 °C-dark in a plant growth chamber. At the stage of fourth true leaves, 10 healthy and uniformly growing plants were selected from each cotton accession and were divided into two groups. These groups individually were watered either with 100 ml of NaCl solution containing a NaCl content of 0.4 % by weight of the sand in the pot as the salinization treatment or with the 100 ml of pure water as the control. The third and fourth leaves from the salt-treated and control plants were collected on the tenth day after the beginning of salinization treatment and stored at −80 °C for later use.
To test the salt tolerance level of the three cotton accessions, germination ratios under salt treatment and control were tested with three replicates for each cotton accession. Relative germination ratio of each cotton accession was calculated to measure the salt tolerance, with the formulation of relative germination ratio (%) = germination ratio under salt treatment/germination ratio of control × 100 (Zhang et al. 2010).
DNA isolation and MSAP assay
DNA extraction was performed according to Paterson et al. (1993). RNA in the DNA was digested with the final concentration of 20 μg/ml RNase for 30 min at room temperature. MSAP detection was adapted from Xu et al. (2000), who modified the protocol for AFLP technique described by Vos et al. (1995), using the isoschizomers HpaII and MspI instead of MseI as the frequent cutter while the rare cutter EcoRI was unchanged. First, several primers were used to identify the robustness of the three biological replicates for each sample; the results indicated identical MSAP bands among three replicates (data not shown). These replicates were then mixed to provide a pool sample for the following MSAP analysis. The sequence information for the adapters and primers of pre-amplification and selective amplification are provided in supplemental Table 1 (Table S1).
Two sets of digestion reactions were performed simultaneously. Approximately 0.25 μg of cotton genomic DNA were digested with 10 units each of EcoRI and HpaII (Promega, USA) in a final volume of 25μl of the appropriate standard buffer for 8 h at 37 °C, and then the enzymes were deactivated by placing the sample at 70 °C for 15 min. In the second reaction, the same amount of cotton genome DNA was digested with EcoRI and MspI under the same reaction conditions. The digested fragments were ligated to the adapters in the ligation reaction mixture containing ligase buffer and T4 DNA ligase (Promega, USA), and incubated at 18 °C overnight (about 13 h). The ligation mixture was diluted at 1:5 (v/v) with Tris–EDTA (TE), and used as the template for the pre-selective amplification with EcoRI+A and HpaII/MspI+T primers.
The PCRs were performed as follows: pre-denaturation at 94 °C for 5 min; 20 cycles of 30 s denaturation at 94 °C, 1 min annealing at 56 °C, and 1 min extension at 72 °C; 7 min extension at 72 °C; and finally a 10 °C hold. The pre-amplification products were diluted 20-fold (v/v) with TE buffer and used as the template for the selective amplification reaction. In this step, EcoRI and HpaII/MspI primers with two additional selective nucleotides were used. The selective PCR was performed in a final volume of 10 μl; according to the protocol of Xu et al. (2000). The PCR reaction was performed for 36 cycles with the following cycle profile (Vos et al. 1995): pre-denaturation at 94 °C for 5 min; 13 touchdown cycles (30 s denaturation at 94 °C, 30 s annealing at 65 °C, which was subsequently reduced each cycle by 0.7 °C for the next 12 cycles, and 1 min extension at 72 °C); 23 cycles of 30 s denaturation at 94 °C, 30 s annealing at 56 °C, and 1 min extension at 72 °C; 7 min extension at 72 °C; and finally a 10 °C hold. The products of PCR were separated with CE on GenomeLab CEQ/GeXP genetic analysis system according to the manual (Beckman, USA). The scored MSAP bands were transformed into a binary character matrix, using “0”and “1”to indicate the absence and presence, respectively, of particular sites.
For each single DNA sample, the MSAP patterns resulted from the digestions with the isoschizomers were divided into the following four types: type I bands, present only for EcoRI/MspI (0,1), which represent the case of full methylation of internal cytosine of 5′-CCGG on the two DNA strands; type II bands, present only for EcoRI/HpaII (1,0), which represent the hemimethylated state of 5′-CCGG sites due to methylation in one DNA strand (external cytosine or both external and internal cytosines of 5′-CCGG) but not in its complementary strand; type III bands, absent from both enzyme combinations (0, 0), which represent full methylation of the two strands where at least one external cytosine of 5′-CCGG was methylated; type IV bands, present for both enzyme combinations (1, 1), which represent the case of no methylation, or the hemimethylated state of 5′-CCGG sites due to methylation of internal cytosine of 5′-CCGG on one DNA strand but not on its complementary strand, and type IV bands were usually treated as no methylation in analysis although it will underestimate actual methylation level. Type I, II, IV can be identified according to any single DNA sample; however, type III (0, 0) can only be detected when comparing to other samples. The percentage of polymorphic MSAP bands can be calculated using the following formula:
Cloning and BLAST search of differentially methylated fragments
The surplus samples used for CE were separated on 10 % conventional polyacrylamide gels so as to clone the interesting bands. DNA recovery, vector ligation, and transformation of DH5a Escherichia coli competent cells were conducted as described by Guo et al. (2003). At least three positive clones from each amplified fragment were simultaneously sequenced by Shanghai Generay Biotech Co., Ltd, China. Homology search was performed on the National Center for Biotechnology Information BLAST server (USA) using the nucleotide sequence for each cloned sequence.
RNA extraction and real-time quantitative PCR (RT-qPCR)
Total RNA was extracted with the EASY spin RNA plant kit (Aidlab, Beijing, China) according to the manufacturer’s instructions. RNA was extracted from 100 mg of leaf tissue sampled at the same time-point that DNA methylation samples were taken for analysis and then stored at −80 °C. RNAs were then quantitated by UV spectrophotometry and cDNA was constructed using RNA samples and the M-MLV reverse transcriptase kit (Invitrogen). The RT-qPCR procedure was based on the iCycler iQTM real-time PCR detection system (Bio-Rad), following the manufacturer’s instructions. Primers were designed using the Primer 3 software (http://www.simgene.com/Primer3) and then synthesized by Takara Biotech Co., Ltd (Dalian, China). The sequences of primers are described in supplemental Table 2 (Table S2). For normalization, the G. hirsutum glyceraldehyde-3-phosphate dehydrogenase (GhGAPDH, GenBank accession numbers: FJ415206) was used as an endogenous control. All PCR reactions were performed in triplicate, and the specificity of the reaction was detected by melting-curve analysis at the dissociation stage. Comparative quantification of each target gene was performed based on cycle threshold (CT) normalized to GhGAPDH using the ∆∆CT method.
Results
Salt tolerance level of different cotton accessions
The salt-tolerance levels of the three cotton accessions were listed in Table 1. The results showed that CCRI 12 was sensitive to salt stress, with the relative germination ratio of 31.34 %. Zhong 07 and CCRI 35 had significantly higher salt tolerance level than that of CCRI 12, with relative germination ratio of 79.51 and 76.23 % respectively (Table 1; Zhang et al. 2010).
DNA methylation level of different cotton accessions
The DNA methylation level of the three cotton accessions were identified with MSAP based on CE (Table 2). MSAP based on CE detected 3090 CCGG sites in CCRI 35 under salt treatment and the control (Table 2) of which 2665 (86.2 %) and 1986 (64.3 %) sites were methylated under the salt treatment and the control respectively. The average number of sites detected per primer combination for the salt treatment and the control were 83.3 and 62.1 respectively. The methylated site number under salt stress was significantly higher than that of control (P < 0.01). For the type III methylation, 1668 and 356 methylated sites that were detected under the salt treatment and control respectively, the methylation level under the salt treatment is significantly higher than that of control (P < 0.01). For the type I methylation, 339 and 838 methylated sites were detected under the salt treatment and control respectively; the methylation level under the salt treatment is significantly lower than that of control (P < 0.01). MSAP based on CE detected 2690 CCGG sites in Zhong 07 under the salt treatment and the control treatment (Table 2) of which 2280 (84.8 %) and 1920 (71.4 %) sites were methylated under the salt treatment and the control respectively. The average sites detected per primer combination were 71.3 and 60 respectively, and the methylated site number under the salt treatment was significantly higher than that of control (P < 0.01). For the type III methylation, 1191 and 447 methylated sites were detected under the salt treatment and control respectively; the methylation level under the salt treatment was significantly higher than that of control (P < 0.01). For the type I methylation, 410 and 977 methylated sites were detected under the salt treatment and control respectively; the methylation level under salt stress was significantly lower than that of control (P < 0.01). In total, 2173 CCGG sites were detected in CCRI 12 (Table 2) of which 1479 (68.1 %) and 1648 (75.8 %) sites were methylated under the salt treatment and the control, respectively. The average number of sites detected per primer combination were 46.2 and 51.5 under the salt treatment and the control, respectively, with no significant difference existed between salt stress and control.
Variation of DNA methylation status of cotton accessions under salt stress
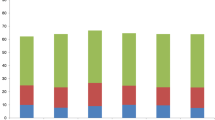
The methylation level compared between the salt treatment and the control can be divided into four patterns which include A, B, C, and D (Fig. 1; Table 3), and furthermore these four patterns can be divided into 15 sub-patterns (Table 3). Pattern A indicates that the methylation status is the same between the salt treatment and the control without changes. Pattern B indicates that DNA methylation level decreases under the salt treatment, or hypomethylation. Pattern C indicates that DNA methylation level increased under the salt treatment, or hypermethylation. Pattern D indicates that DNA methylation status changed between the salt treatment and the control, but one cannot tell the difference between treatment and control in the methylation level.
The detailed composition of different patterns and their percentages are listed in Table 3. Especially, pattern B is composed of five sub-patterns with 14, 21, and 46.5 % of total sites belonging to this pattern in CCRI 35, Zhong 07, and CCRI 12 respectively; pattern C is also composed of five sub-patterns, and 66, 55.1, and 20.5 %of total sites belonged to this pattern in CCRI 35, Zhong 07, and CCRI 12, respectively. The results showed that in cotton accessions CCRI 35 and Zhong 07, hypermethylation that happened under salt stress as compared to the control occurs significantly more often (P < 0.01) than hypomethylation (Table 3); whereas in CCRI 12, hypomethylation happened significantly more often (P < 0.01) under salt stress as compared to the control than hypermethylation (Table 3).
The four banding patterns were also compared between different cotton accessions (Fig. 1). For pattern B, CCRI 35 and Zhong 07 had no significant differences, whereas their loci numbers were significantly lower than that of CCRI 12. For pattern C, CCRI 35 and Zhong 07 also had no significant differences, whereas their loci numbers were significantly higher than that of CCRI 12 (Fig. 1).

Variation of methylation banding pattern of the three cotton accessions A Same methylation level between control and salt stress; B methylation level decreased under salt stress (hypomethylation); C methylation level increased under salt stress (hypermethylation); D methylation pattern changed. The small letters a and b differences at level of P < 0.05 between different varieties. Green pattern CCRI35, red–grey pattern Zhong 07, blue pattern CCRI 12
Differentially methylated DNA sequences and gene homology
Cloning and BLAST search analysis of methylated DNA sequences found 12 sequences that have high similarity to 10 genes or non-coding regions (Table 4; Table S3 in supplementary material). The homologous genes were related to transducin, transcription factor, calcium-binding protein, ATPase, plasma-membrane choline transporter, Leucine-rich receptor-like protein kinase family, cytokinin oxidase, potassium channel and so on. Eight of the 12 sequences had the same DNA methylation status between salt stress and the control. Two sequences experienced hypermethylation and another two sequences experienced hypomethylation under salt stress (Table 4).
Gene expression analysis of selected MSAP fragments
Three interesting MSAP fragments, which will be important to our further research, was selected and analyzed for their transcript level by means of RT-qPCR. Transcript abundance was assessed in all treated samples and normalized against controls. The sequence YF09 homologous to a MYB transcription factor experienced hypomethylation under salt stress in CCRI 35 (Table 4) and was significantly up-regulated under salt stress (Fig. 2a). Meanwhile, it was mildly up-regulated under salt stress in Zhong 07 and significantly up-regulated under salt stress in CCRI 12 (Fig. 2a). The sequence YF25 homologous to Gossypium hirsutum clone de-Met-HMTGC-EACG genome sequence maintained the same methylation level after salt treatment in CCRI 12 (Table 4) and its expression level did not vary significantly after salt treatment in CCRI 12 (Fig. 2b). However, it was significantly down-regulated under salt treatment in CCRI 35 and Zhong 07 (Fig. 2b). The sequence YF57 related to cytokinin oxidase maintained the same methylation level between salt treatment and control in Zhong 07 (Table 4); its expression level did not vary significantly after salt treatment in Zhong 07 and CCRI 12. However, it was significantly up-regulated under salt treatment in CCRI 12 (Fig. 2c).

The expression of genes detected by RT-qPCR in the three cotton accessions. The transcript levels were normalized to that of GhGAPDH, and the level of each gene in the control samples was set at 1.0. Error bars mean value (±SE, n = 3), asterisk(s) significant differences between salt treatment and control at P < 0.05 (*). a GU324346.1, b EF159966.1, c XM_007012355.1. Pink control, blue salt treatment
Discussion
DNA methylation and salt stress
DNA methylation is a modification that occurs at the fifth carbon position of a cytosine ring. DNA methylation plays a role in diverse cellular activities, such as carcinogenesis, gene silencing, genomic imprinting, chromatin remodeling, dosage compensation, DNA replication timing, and disease defense and is known to be meiotically as well as mitotically inherited in plants (Keyte et al. 2006). In both plants and animals, cytosine is primarily methylated in the CG dinucleotide context, and methylation in the 5′ portion of the gene and the 3′ portion may inhibit gene expression (Grativol et al. 2012), consequently, the genes with very low level of expression are most likely to be methylated; in contrast, the most expressed genes can be methylated with much less probability (Zemach et al. 2010). Regulation of gene expression is important to plants’ tolerance for stresses. In this context, methylation is gaining interest because it may regulate the expression of genes that have a key role in acclimation responses. In addition, it is well known that transposons are directly regulated by methylation and that they may have an important role in plant response to stress. Some epigenetic states are established by transiently expressed or transiently activated factors that respond to environmental stimuli (Bonasio et al. 2010).
In many cases, a global demethylation of genomic DNA occurs in response to abiotic stress, suggesting that there might be a close correlation between demethylation and gene expression. Contrastingly, sometimes hypermethylation also occurs in response to stress. These results imply that the DNA methylation pattern and alteration are related to the abiotic stress response in plants but that the association is obviously different between plant species, and there seems to be a relationship between DNA-methylation types and stress responses. For instance, wild-type tobacco plants exposed to aluminum, salt, paraquat, and cold stress showed a selective decrease of CG methylation in the coding region of the glycerophosphodiesterase-like protein gene (Choi and Sano 2007). Tobacco plants infected with tobacco mosaic virus showed strong CG hypomethylation at the Leucine-rich repeat region of the gene for resistance to tobacco mosaic virus (Boyko et al. 2007). It is noted that biotic stresses such as pathogenic infection can lead to two contrasting effects on the levels of methylation in plants: hypermethylation on the genome-wide level and hypomethylation of resistance-related genes (Peng and Zhang 2009). Both phenomena may contribute to the adaptation of plants to stress. Previous research found that chromium stress can promote methylation in radish (Yang et al. 2007) and oilseed rape (Labra et al. 2004) but induces a decrease in methylation in clover and hemp (Aina et al. 2004). This suggests that different methylation mechanisms for heavy-metal resistance exist in different plant species.
Under salt stress conditions, Wang et al. (2011b) found that demethylation of genes was an active epigenetic response to salt stress in roots at the rice seedling stage; whereas Kovarik et al. (1997) pointed out that tobacco cell cultures exposed to osmotic stress showed CHG hypermethylation without changes in CG methylation in two heterochromatic sites, and Dyachenko et al. (2006) noticed that the level of CHG methylation of the halophyte Mesembryanthemum crystallinum increased in a satellite DNA on switching-over of C3-photosynthesis to the crassulacean acid metabolism pathway of carbon dioxide assimilation. In cotton research, it was found that demethylation positively contributed to salt tolerance. For instance, Li et al. (2009) found that the level of global DNA methylation in cotton roots decreased after salt stress, and there was a significantly negative correlation between salt concentration and DNA methylation level. Zhao et al. (2010) suggested that the demethylation positively contributed to salt tolerance and the hypermethylation had negative effect on salt tolerance in cotton. Cao et al. (2011) found that demethylation occurred more than methylation in cotton roots and leaves under salt and alkali stress.
In our current research, DNA methylation was detected for different cotton accessions under salt stress. The results showed that variations of DNA methylation happened in the three cotton accessions under salt stress, including hypermethylation, hypomethylation, and other patterns. For the two salt-tolerant cotton accessions CCRI 35 and Zhong 07, the methylation level significantly increased under salt stress (Table 2), whereas for the salt-sensitive cotton accession CCRI 12, there was no significant changes between salt stress and control, and the methylation level under salt stress was even lower than that of control (Table 2). It is interesting to further find out that for the two salt-tolerant cotton accessions, the numbers of type I methylation patterns under salt stress were significantly lower than that of control, whereas the number of type III methylation pattern under salt stress was significantly higher than that of control.; on the contrary, the salt-sensitive cotton accession CCRI 12 had a significant opposite tendency (Table 2). The different varying tendency of specific methylation patterns between salt-tolerant and salt-sensitive cotton accessions indicate that specific methylation patterns might also be related to salt tolerance in cotton.
When comparing the variance of DNA methylation patterns, hypermethylation under salt stress occurred most often with statistical significance in the two salt-tolerant cotton accessions CCRI 35 and Zhong 07, whereas hypomethylation under salt stress occurred most often with statistical significance in the salt-sensitive cotton accessions CCRI 12 (Table 3). In addition, for pattern B or hypomethylation pattern, salt-tolerant accessions CCRI 35 and Zhong 07 had no significant differences, whereas their loci numbers were significantly lower than that of the salt-sensitive accession CCRI 12; for pattern C or hypermethylation, CCRI 35 and Zhong 07 also had no significant differences, whereas their loci numbers were significantly higher than that of CCRI 12 (Fig. 1). The significant increase of global DNA methylation level under salt stress in salt-tolerant accessions but not in salt-sensitive accessions suggests that salt-tolerant cotton accessions have a mechanism of increasing methylation level that in turn affects the response to salt stress.
The different results in our research compared to other researches (Li et al. 2009; Zhao et al. 2010; Cao et al. 2011) might be due to the adoption of different cotton materials or different cotton tissues, whereas DNA methylation is tissue-specific (Angers et al. 2010): e.g., roots have different microenvironment of rhizosphere as compared to leaf surroundings, and also there exist functional differentiation between tissues as leaf tissues are more complex and highly differentiated than root tissues (Mastan et al. 2012). Osabe et al. (2014) also found that significantly different levels of DNA methylation existed between different cotton developmental stages or tissues, suggesting the importance of epigenetic regulation in creating phenotypic diversity. On the other hand, the variation in methylation is dynamic. The flexibility and dynamic features of DNA methylation modifications require us to accurately determine the time points at which observations are made. Contrary conclusions may result from errors of just a few hours. Observation at multiple time points and repeated analysis under several rounds of stress are both needed to determine whether the variations in methylation are reversed after removal of the stress (Peng and Zhang 2009).
Genes responding to salt stress in cotton
Cloning and BLAST search analysis of methylated DNA sequences found 14 sequences that have high similarity to genes or non-coding regions, and the known genes were related to transducin, transcription factor, calcium-binding protein, ATPase, plasma-membrane choline transporter, leucine-rich receptor-like protein kinase family, cytokinin oxidase, potassium channel and so on (Table 4). It is not surprise that 10 of the 14 sequences were homologous to genes of Theobroma cacao, since molecular phylogenetic analyses suggested that the common ancestor of G. arboreum and G. raimondii having diverged from T. cacao 18–58 million years ago (Wikström et al. 2001).
The sequence YF24 was homologous to an ATP synthase gene, whereas the sequence YF59 was homologous to a potassium channel gene. Due to the essential role of K+ in the plant as a co-factor for many cytosolic enzymes, Na+ toxicity results from its competition with K+ at binding sites, and can inactivate the enzymes active sites and essential cellular functions. Highly salt-resistant species are able to maintain low cytosolic free Na+/K+ and Na+/Ca2+ ratios (Sun et al. 2010). The ability to control Na+/K+ and Na+/Ca2+ homeostasis in salinity resistant plants is usually associated with a higher plasma membrane H+-ATPase activity and proposed to be involved in salt tolerance (Sun et al. 2010; Morgan et al. 2014). In our current research, the ATP synthase gene and the potassium channel gene maintained the same methylation level after salt stress in CCRI 35 and Zhong 07 respectively (Table 4), so their expression should not be inhibited by DNA methylation, which might be part of the reason that CCRI 35 and Zhong 07 are salt tolerant and can maintain normal growth after salt stress in a certain period.
Cytokinins (CK) are master regulators of plant growth and development, and were recently shown to control plant adaptation to salt stress (Nishiyama et al. 2012). Nishiyama et al. (2011, 2012) suggest that reduction of bioactive CK levels induced changes in gene expression of many regulatory and functional genes, leading to improved salt and drought stress tolerance. All CK-deficient plants with reduced levels of various CKs exhibited a strong stress-tolerant phenotype that was associated with increased cell membrane integrity and abscisic acid (ABA) hypersensitivity rather than stomatal density and ABA-mediated stomatal closure (Nishiyama et al. 2012). In plants, the key enzymes involved in CK metabolism are adenosine phosphate-isopentenyltransferases and CK oxidases/dehydrogenases, which catalyzed irreversibly degradation of CK (Hirose et al. 2008). Therefore, CK oxidases are important players in regulating CK concentrations and thereby influence plant growth and development. We found that the sequence YF57 related to Theobroma cacao cytokinin oxidase maintained the same DNA methylation level in Zhong 07 between salt treatment and control (Table 4). RT-qPCR results showed that its expression level was higher and did not vary significantly after salt treatment in CCRI 35 and Zhong 07, the two salt-tolerant cotton accessions; whereas its expression was lower and significantly up-regulated after salt treatment in the salt-sensitive cotton accession, CCRI 12 (Fig. 2c). It is deduced that the salt-sensitive cotton accession CCRI 12 responded more quickly to salt stress and the overexpression of cytokinin oxidase will help reduce the cytokinin level, which should be part of its defense systems responding to salt stress.
In our research, the sequences YF09 experienced hypermethylation under salt stress in CCRI 35 with the methylation pattern of B4, which is related to a MYB transcription factor GU324346.1 (Table 4). Key components that control and modulate stress adaptive pathways include transcription factors ranging from bZIP, AP2/ERF, and MYB proteins to general transcription factors (Golldack et al. 2011). Plant MYB transcription factor genes that execute positive or negative regulation of diverse types of stress responses have been reported. For instance, several MYB transcription factors have been reported to involve plant response to drought stress; also, both AtMYB2 and AtMYB44 were identified as regulators of ABA-dependent salt and drought stress responses (Gao et al. 2014). Heterologous expression of the chrysanthemum R2R3-MYB transcription factor CmMYB2 enhanced tolerance to drought and saline stresses (Shan et al. 2012). An Arabidopsis R2R3-MYB transcription factor AtMYB20 negatively regulates type 2C serine/threonine protein phosphatases to enhance salt tolerance (Cui et al. 2013). GhWRKY39, a member of the WRKY transcription factor family in cotton, has a positive role in disease resistance and salt stress tolerance (Shi et al. 2014). Some reports also indicated that transcription factors may be negatively involved in plant adaptive response to stresses. Gao et al. (2014) found that a R2R3-MYB transcription factor AtMYB20 can be differently regulated by abiotic stresses: while AtMYB20 was induced by high levels of NaCl, its expression was suppressed by desiccation and cold. In a transcriptome analysis of responsive genes to multiple abiotic stresses in cotton, Zhu et al. (2013) found that WRKY family was the most highly expressed transcription factor, while ERF was the most important repressed transcription factor under abiotic stress conditions in cotton. Here in our research, RT-qPCR validated that the MYB transcription factor gene GU324346.1 was significantly up-regulated under salt stress in CCRI 35 and CCRI 12, and mildly up-regulated in Zhong 07 (Fig. 2a), indicating that its overexpression is positively related to salt tolerance.
The sequence YF25 homologous to Gossypium hirsutum clone de-Met-HMTGC-EACG genome sequence maintained the same methylation level after salt treatment in CCRI 12 (Table 4) and its expression level did not vary significantly after salt treatment in salt-sensitive CCRI 12 (Fig. 2b). However, it was significantly down-regulated under salt treatment in the salt-tolerant accessions, CCRI 35 and Zhong 07 (Fig. 2b), consequently, although its function is not clear so far, it might be of special importance to cotton salt tolerance. It is interesting to find that three sequences cloned were related to genes controlling a Theobroma cacao leucine-rich receptor-like protein kinase family protein in CCRI 35, Zhong 07 and CCRI 12 respectively (Table 4), and they all maintained the same methylation level after salt treatment. It is reported that a receptor-like kinase gene (GbRLK) from Gossypium barbadense enhances salinity and drought-stress tolerance in Arabidopsis (Zhao et al. 2013), so we deduce that the stable expression of this gene might contribute to salt stress in cotton. Our future research will perform function validation experiments on these interesting genes and try to shed light to further salt tolerance mechanism in cotton.
References
Aina R, Sgorbati S, Santagostino A, Labra A, Ghiani A, Citterio S (2004) Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp. Physiol Plantarum 121:472–480
Angers B, Castonguay E, Massicotte R (2010) Environmentally induced phenotypes and DNA methylation: how to deal with unpredictable conditions until the next generation and after. Mol Ecol 19:1283–1295
Bilichak A, Ilnystkyy Y, Hollunder J, Kovalchuk I (2012) The progeny of Arabidopsis thaliana plants exposed to salt exhibit changes in DNA methylation, histone modifications and gene expression. PLoS ONE 7:e30515
Bonasio R, Tu S, Reinberg D (2010) Molecular signals of epigenetic states. Science 330:612–616
Boyko A, Kathiria P, Zemp FJ, Yao Y, Pogribny I, Kovalchuk I (2007) Transgenerational changes in the genome stability and methylation in pathogen-infected plants. Nucleic Acids Res 35:1714–1725
Cao DH, Gao X, Liu J, Kimatu JN, Geng SL, Wang XP, Zhao J, Shi DC (2011) Methylation sensitive amplification polymorphism (MSAP) reveals that alkali stress triggers more DNA hypomethylation levels in cotton (Gossypium hirsutum L.) roots than salt stress. Afr J Biotechnol 10:18971–18980
Choi CS, Sano H (2007) Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol Genet Genomics 277:589–600
Cui MH, Yoo KS, Hyoung S, Nguyen HTK, Kim YY, Kim HJ, Ok SH, Yoo SD, Shin JS (2013) An Arabidopsis R2R3-MYB transcription factor, AtMYB20, negatively regulates type 2C serine/threonine protein phosphatases to enhance salt tolerance. FEBS Lett 587:1773–1778
Dyachenko OV, Zakharchenko NS, Shevchuk TV, Bohnert HJ, Cushman JC, Buryanov YI (2006) Effect of hypermethylation of CCWGG sequences in DNA of Mesembryanthemum crystallinum plants on their adaptation to salt stress. Biochemistry (Moscow) 71:461–465
Flowers TJ, Yeo AR (1995) Breeding for salinity resistance in crop plants: where next? Aust J Plant Physiol 22:875–884
Gao S, Zhang YL, Yang L, Song JB, Yang ZM (2014) AtMYB20 is negatively involved in plant adaptive response to drought stress. Plant Soil 376:433–443
Golldack D, Lüking I, Yang O (2011) Plant tolerance to drought and salinity: stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep 30:1383–1391
Grativol C, Hemerly AS, Ferreira PCG (2012) Genetic and epigenetic regulation of stress responses in natural plant populations. BBA-Gene Regul Mech 1819:176–185
Guo WZ, Zhang TZ, Sheng XL, John Y, Kohel RJ (2003) Development of SCAR marker linked to a major QTL for high fiber strength and its molecular marker assisted selection in Upland cotton. Crop Sci 6:2252–2256
Hirose N, Takei K, Kuroha T, Kamada-Nobusada T, Hayashi H, Sakakibara H (2008) Regulation of cytokinin biosynthesis, compartmentalization and translocation. J Exp Bot 59:75–83
Jiang YR, Lv YJ, Zhu SJ (2006) Advance in studies of the mechanism of salt tolerance and controlling of salt damage in Upland cotton. Cotton Sci 18:248–254
Keyte AL, Percifield R, Liu B, Wendel JF (2006) Infraspecific DNA methylation polymorphism in cotton (Gossypium hirsutum L.). J Hered 97:444–450
Kimatu JN, Diarso M, Song CD, Agboola RS, Pang JS, Qi X, Liu B (2011) DNA cytosine methylation alterations associated with aluminium toxicity and low pH in Sorghum bicolor. Afr J Agr Res 6:4579–4593
Kovarik A, Koukalova B, Bezdek M, Opatrny Z (1997) Hypermethylation of tobacco heterochromatic loci in response to osmotic stress. Theor Appl Genet 95:301–306
Labra M, Grassi F, Imazio S, Fabio TD, Citterio S, Sgorbati S, Agradi E (2004) Genetic and DNA-methylation changes induced by potassium dichromate in Brassica napus L. Chemosphere 54:1049–1058
Li XL, Lin ZX, Nie YC, Guo XP, Zhang XL (2009) MSAP analysis of epigenetic changes in cotton (Gossypium hirsutum L.) under salt stress. Acta Agro Sinica 35:588–596
Lukens LN, Zhan SH (2007) The plant genome’s methylation status and response to stress, implications for plant improvement. Curr Opin Plant Biol 10:317–322
Mahajan S, Tuteja N (2005) Cold, salinity and drought stresses: an overview. Arch Biochem Biophys 444:139–158
Mastan SG, Rathore MS, Bhatt VD, Yadav P, Chikara J (2012) Assessment of changes in DNA methylation by methylation-sensitive amplification polymorphism in Jatropha curcas L. subjected to salinity stress. Gene 508:125–129
McClelland M, Nelson M, Raschke E (1994) Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res 22:3640–3659
Morgan SH, Maity PJ, Geilfus CM, Lindberg S, Mühling KH (2014) Leaf ion homeostasis and plasma membrane H+-ATPase activity in Vicia faba change after extra calcium and potassium supply under salinity. Plant Physiol Biochem 82:244–253
Nishiyama R, Watanabe Y, Fujita Y, Le DT, Kojima M, Werner T, Vankova R, Yamaguchi-Shinozaki K, Shinozaki K, Kakimoto T, Sakakibara H, Schmülling T, Trana LSP (2011) Analysis of cytokinin mutants and regulation of cytokinin metabolic genes reveals important regulatory roles of cytokinins in drought, salt and abscisic Acid responses, and abscisic acid biosynthesis. Plant Cell 23:2169–2183
Nishiyama R, Le DT, Watanabe Y, Matsui A, Tanaka M, Seki M, Yamaguchi-Shinozaki K, Shinozaki K, Tran LS (2012) Transcriptome analyses of a salt-tolerant cytokinin-deficient mutant reveal differential regulation of salt stress response by cytokinin deficiency. PLoS ONE 7:e32124
Osabe K, Clement JD, Bedon F, Pettolino FA, Ziolkowski L, Llewellyn DJ, Finnegan EJ, Wilson IW (2014) Genetic and DNA methylation changes in cotton (Gossypium) genotypes and tissues. PLoS ONE 9:e86049
Paterson AH, Brubaker CL, Wendel JF (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol Biol Rep 11:122–127
Peng H, Zhang J (2009) Plant genomic DNA methylation in response to stresses: potential applications and challenges in plant breeding. Prog Nat Sci 19:1037–1045
Reyna-López GE, Simpson J, Ruiz-Herresa J (1997) Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol Gen Genet 253:703–710
Shan H, Chen S, Jiang J, Chen F, Chen Y, Gu C, Li P, Song A, Zhu X, Gao H, Zhou G, Li T, Yang X (2012) Heterologous expression of the Chrysanthemun R2R3-MYB transcription factor CmMYB2 enhances drought and salinity tolerance, increases hypersensitivity to ABA and delays flowering in Arabidopsis thaliana. Mol Biotechnol 51:160–173
Shan XH, Wang XY, Yang G, Wu Y, Su SZ, Li SP, Liu HK, Yuan YP (2013) Analysis of the DNA methylation of Maize (Zea mays L.) in response to cold stress based on methylation-sensitive amplified polymorphisms. J Plant Biol 56:32–38
Shi WN, Liu DD, Hao LL, Wu CA, Guo XQ, Li H (2014) GhWRKY39, a member of the WRKY transcription factor family in cotton, has a positive role in disease resistance and salt stress tolerance. Plant Cell Tissue Organ Cult 118:17–32
Sun J, Wang MJ, Ding MQ, Deng SR, Liu MQ, Lu CF, Zhou XY, Shen X, Zheng XJ, Zhang ZK, Song J, Hu ZM, Xu Y, Chen SL (2010) H2O2 and cytosolic Ca2+ signals triggered by the PM H+-coupled system mediate K+/Na+ homeostasis in NaCl-stressed Populus euphratica cells. Plant Cell Environ 33:943–958
Tan MP (2010) Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol Biochem 48:21–26
Türkana I, Demiral T (2009) Recent developments in understanding salinity tolerance. Environ Exp Bot 67:2–9
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Fijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Wang WS, Pan YJ, Zhao XQ, Dwivedi D, Zhu LH, Ali J, Fu BY, Li ZK (2011a) Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J Exp Bot 62:1951–1960
Wang WS, Zhao XQ, Pan YJ, Zhu LH, Fu BY, Li ZK (2011b) DNA methylation changes detected by methylation-sensitive amplified polymorphism in two contrasting rice genotypes under salt stress. J Genet Genomics 38:419–424
Wikström N, Savolainen V, Chase MW (2001) Evolution of the angiosperms: calibrating the family tree. Proc Biol Sci 268:2211–2220
Xiong LZ, Xu CG, Saghai Maroof MA, Zhang QF (1999) Patterns of cytosine methylation pattern in an elite rice hybrid and its parental lines detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet 261:139–446
Xu ML, Li XQ, Korban SS (2000) AFLP-based detection of DNA methylation. Plant Mol Biol Rep 18:361–368
Xu Q, Sun DX, Zhang Y (2005) F-MSAP: a practical system to detect methylation in chicken genome. Chin Sci Bull 50:2039–2044
Yang JL, Liu LW, Gong YQ, Huang DQ, Wang F, He LL (2007) Analysis of genomic DNA methylation level in radish under cadmium stress by methylation sensitive amplified polymorphism technique. J Plant Physiol Mol Biol 33:219–226
Zemach A, McDaniel IE, Silva P, Zilberman D (2010) Genome-wide evolutionary analysis of eucaryotic DNA methylation. Science 328:916–919
Zhang LN, Ye WW, Wang JJ, Fan BX (2010) Studies of salinity-tolerance with SSR markers on G. hirsutum L. Cotton Sci 22:175–180
Zhao YL, Yu SX, Ye WW, Wang HM, Wang JJ, Fan BX (2010) Study on DNA cytosine methylation of cotton (Gossypium hirsutum L.) genome and its implication for salt tolerance. Agric Sci China 9:783–791
Zhao J, Gao Y, Zhang Z, Chen T, Guo W, Zhang T (2013) A receptor-like kinase gene (GbRLK) from Gossypium barbadense enhances salinity and drought-stress tolerance in Arabidopsis. BMC Plant Biol 13:110
Zhu JK (2001) Plant salt tolerance. Trends Plant Sci 6:66–71
Zhu YN, Shi DQ, Ruan MB, Zhang LL, Meng ZH, Liu J, Yang WC (2013) Transcriptome analysis reveals crosstalk of responsive genes to multiple abiotic stresses in cotton (Gossypium hirsutum L.). PLoS ONE 8:e80218
Acknowledgments
Research supported by the Natural Science Foundation of Jiangsu Province of China (BK20131204, BK20130429), the National Natural Science Foundation of China (31000729), the State Key Laboratory of Cotton Biology Open Fund (CB2015A09, CB2013A12), the State Foundation for Studying Abroad (2015), the National Practice Innovation Training Program Projects for College Students (2015).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below are the links to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, B., Fu, R., Zhang, M. et al. Analysis of methylation-sensitive amplified polymorphism in different cotton accessions under salt stress based on capillary electrophoresis. Genes Genom 37, 713–724 (2015). https://doi.org/10.1007/s13258-015-0301-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-015-0301-6