
Abstract
Overproduction of livestock manures with unpleasant odors causes significant environmental problems. The microbial fermentation bed (MFB) system is considered an effective approach to recycling utilization of agricultural byproducts and pig manure (PM). To gain a better understanding of bacterial communities present during the degradation of PM in MFB, the PM bacterial community was evaluated at different fermentation stages using 16S rRNA high throughput sequencing technology. The heatmap plot clustered five samples into short-term fermentation stage of 0–10 days and long-term fermentation stage of 15–20 days. The most abundant OTUs at the phylum level were Firmicutes, Actinobacteria and Proteobacteria in the long-term fermentation stage of PM, whereas Firmicutes, Bacteroidetes, and Proteobacteria predominated in the short-term fermentation stage of PM. At the genus level, organic degradation strains, such as Corynebacterium, Bacillus, Virgibacillus, Pseudomonas, Actinobacteria, Lactobacillus, Pediococcus were the predominate genera at the long-term fermentation stage, but were found only rarely in the short-term fermentation stage. C/N ratios increased and the concentration of the unpleasant odor substance 3-hydroxy-5-methylisoxazole (3-MI) decreased with prolonged period of fermentation. Redundancy analysis (RDA) demonstrated that the relative abundance of Firmicutes, Actinobacteria, Acidobacteria and Proteobacteria had a close relationship with degradation of 3-MI and increasing C/N ratio. These results provide valuable additional information about bacterial community composition during PM biodegradation in animal husbandry.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
The large quantities of animal manures produced in the livestock industry lead to serious environmental problems (Ni et al. 2012). Pig manure (PM)—a major source of livestock manure—causes extensive concern (Chadwick et al. 2015). The disposal of PM attracts public attention worldwide. Intensive livestock production is the main cause of the release of 3-hydroxy-5-methylisoxazole (3-MI, CAS: 83–34-1) to the environment. 3-MI (also referred to as skatole) is commonly found in feces and sewage, and is formed by the bacterial degradation of tryptophan (Jensen et al. 1995; Nakai et al. 1999). Skatole has an unpleasant odor, and leads to extensive pollution due to its persistence, mobility and potential health impacts (Deslandes et al. 2001), with 3-MI derivatives being highly toxic to microorganisms and animals, as well as harmful to the pork industry (Arora et al. 2015). There is a need for environmentally sustainable and cost-effective approaches to treatment of PM for pig rearing. A new type of PM treatment facility using a microbial fermentation bed system (MFB system) developed by author’s research team has obtained satisfactory results (Zheng et al. 2011).
The MFB system, also known as the deep-litter-system, breeding pig on litter, or in situ decomposition of PM, comprises deep-bedded groove structures on which the animals are raised on a thick layer of litter (Tam 1995; Morrison et al. 2007; Neumann et al. 2015). A large scale MFB equipped with an automatic control system can raise more than 2000 pigs at a time, and is also a productive system for decomposing PM (Liu et al. 2011). The pig feces are mixed with the litter to decompose, without waste emission or spread of odors (Menalled et al. 2005; Rashad et al. 2010; Karadag et al. 2013; Costa et al. 2015). In large scale MFB on pig farms, microorganisms such as bacteria, fungi and actinomycetes play pivotal roles in the degradation of pig feces and composting process (López-González et al. 2014). Ubiquity in the environment, metabolic versatility, and the ability to adapt to extreme conditions were the major factors influencing the effectiveness of bacteria in compost (Liu et al. 2011; Li et al. 2013). Arthurson (2011) reported that bacterial communities in fresh manure were different from those in manure stored in deep pits. However, the structures of bacteria community of PM in the MFB system remain obscure.
The main objective of this investigation was to gain a better understanding of the dynamics of bacterial communities in PM under in situ fermentation in the MFB system, and provide appropriate information for biodegradation of PM. In the light of the above considerations, bacterial community dynamics at different fermentation stages in the MFB system were evaluated using 16S rRNA high throughput sequencing technology, along with physicochemical analysis of organic carbon, nitrogen, phosphorus, and potassium concentrations, and odor substance (3-MI).
Materials and methods
Sample collection and treatment
The MFB-equipped piggery was located in a farm in Fuqing County, Fujian Province, China. The breeding area of the fermentation bed was 2000 m2, with 1800 pigs. The mattress materials comprised agricultural byproducts, including otherwise worthless mushroom-planted, mash and rice-husk materials. The depth of the mattress was 0.8 m. The temperature and humidity were controlled at 28–32°C and 70–90%, respectively. Generally, it is necessary to plow the surface litters once every 2–3 days, and the pigs’ daily rooting behaviors also help to loosen the litter.
PM at different phases was collected from the MFB by the five-point sampling method. Fresh PM (PM1), and PM at fermentation times of 5 days (PM2), 10 days (PM3), 15 days (PM4) and 20 days (PM5) were collected at depths from the surface down to 20 cm. A part of the samples was pooled and homogenized for detection of bacterial diversity. The other part was prepared for basic litter properties tests. Total nitrogen (N), total carbon ©), phosphorus (P), and potassium (K) concentrations are shown in Table 1.
HPLC analysis
Samples (2 g) were dissolved in 10 mL methanol in 50 mL sterilization tubes, and the mixture was vortexed for 30 s, then transferred to a 40 °C water bath for 20 min, blended every 5 min. After centrifugation at 12,000 g for 15 min, the supernatant was stored at 4 °C prior to HPLC analysis by Agilent Technologies 1100 HPLC (Palo Alto, CA). Standard 3-MI (Sigma, St. Louis, MO) was dissolved in methanol. After passing through a 0.22 μm-pore filter (Millipore, Bedford, MA), 10 μL filtrate was subjected to the reversed phase HPLC C18 column (4.6 × 50 mm, 5 μm, Welch, Shanghai, China). The column was eluted with solutions of acetonitrile and Milli-Q water: 0–3.5 min, 40%; 4.5–9.0 min, 75%; 11.0–15 min, 40% at a flow rate of 1 mL min−1. The eluate was monitored at 270 nm (Dehnhard et al. 1991).
DNA extraction and sequencing
Community genomic DNA from samples was extracted using the cetyltrimethyl ammonium bromide (CTAB) method. The concentration and purity of genomic DNA was monitored by 1% agarose gels. The V4 hypervariable regions of the 16S rRNA genes were amplified using primers 515F (5′-GTG CCA GCM GCC GCG GTA A-3′) and 806R (5′- GGA CTA CHV GGG TWT CTA AT-3′). All PCR were carried out in 30 μL reactions with 15 μL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs, Waltham, MA), including 0.2 μM forward and reverse primers, and 10 ng template DNA. Thermal cycling consisted of initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, and elongation at 72 °C for 30 s, and a final extension at 72 °C for 5 min. All the PCR reactions were done in triplicate, and then an equal volume of 1× loading buffer (contained SYB green) was mixed with the PCR products. Samples with a bright main band between 400 bp and 450 bp were purified with a GeneJET Gel Extraction Kit (Thermo Scientific) for further experiments. The purified PCR amplicons were sequenced using the Illumina Miseq Platform at Novogene Bioinformatics Technology (Beijing, China).
Paired-end reads from the original DNA fragments were merged using FLASH (V1.2.7, http://ccb.jhu.edu/software/FLASH/) to get raw tags. Qualities filtering on the raw tags were performed according to the QIIME (V1.7.0, http://qiime.org/index.html) quality control process. Chimeras were detected and removed using USEARCH software based on the UCHIME algorithm (http://www.drive5.com/usearch/manual/ uchime_algo.html).
Bioinformatics analysis
Sequences with ≥97% similarity were assigned to the same operational taxonomic units (OTUs) by UPARSE software (UPARSE v7.0.1001, http://drive5.com/uparse/). Taxonomic information for each OTU was annotated by the RDP classifier (Version 2.2, http://sourceforge.net/projects/rdp-classifier/). The taxon abundance of each sample was generated into phylum, class, order, family, and genera levels. All analyses from clustering to alpha (within sample) and beta diversity (between samples) were performed with QIIME (Version 1.7.0) and displayed with R software (Version 2.15.3). Weighted UniFrac was employed for principal component analysis (PCA). Hierarchical clustering of samples was completed using UPGMA. Venn diagram, heatmap and redundancy analysis (RDA) were constructed by Hierarchical clustering. The sequences were deposited in the sequence read archive (SRA), under accession numbers SRR4473783, SRR4473830, SRR4473831, SRR4473832 and SRR4473833.
Results
Physicochemical properties of the manure related to the fermentation stages in the MFB system
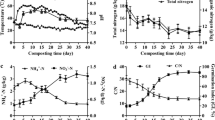
The physicochemical properties of the manure samples are listed in Table 1. Organic matter, total nitrogen and phosphorus were different in PM exposed for different times. The C/N ratios increased from 10.4 (PM1) to 18.4 (PM5) with fermentation of PM. Concentrations of phosphorus and potassium varied among these samples. PM2 and PM3 had the highest content of phosphorus (3.10%) and potassium (2.20%), respectively. PM5 had the lowest content of phosphorus (1.69%) and potassium (1.63%). A gradient levels of skatole appeared in those samples, ranging from 4.2 μg g−1 to 0.7 μg g−1. Among them, PM4 and PM5 had low concentrations of skatole.
Bacterial diversity and richness
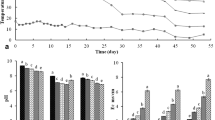
After quality and chimera filtering, a total of 315,761 valid reads were obtained from five samples, with number of sequencing reads ranging from 57,239 (PM4) to 76,417 (PM1) at 3% distance (Table 2). PM1 has the highest value of OTUs (2011), followed by PM2 (1760), PM3 (1758), PM4 (1599) and PM5 (1599). All rarefaction curves tended to approach a plateau, suggesting that the sequencing depth for all samples was reasonable to cover bacterial community diversity (Fig. 1). The alpha diversity including Shannon, Simpson, Chao1 and ACE index could reflect the variation in microbial diversity among those samples. The Shannon indexes of PM1, PM2, PM3, PM4 and PM5 were 8.177, 7.83, 7.761, 5.5576 and 6.311, respectively (Table 2).

Rarefaction curves based on 16S rRNA sequences among the different samples
Taxonomic compositions
Bacterial diversity richness and phylogenetic distribution were analyzed at different stages (Fig. 2). Bacterial community abundance was significantly different among the five PM samples (Fig. 3). The most abundant taxa throughout 31 phyla were Firmicutes, Actinobacteria, Proteobacteria and Bacteroidetes (Fig. 3a). The most abundant OTUs associated with the PM4 library were sequences related to Firmicutes (65.5%), Actinobacteria (22.5%) and Proteobacteria (7.4%). The most abundance sequences in the PM1, PM2 and PM3 library were those related to Firmicutes, Bacteroidetes, and Proteobacteria (Fig. 3a).

Bacterial diversity richness and phylogenetic distribution on phylum (inner circle), class (middle circle), and order level (outer circle)

Relative abundance of operational taxonomic units (OTUs) of predominant bacterial compositions at a phylum and b genus levels in the microbial fermentation bed (MFB) system
The most abundant bacteria in PM4 at the genus level were Lactobacillus (44.9%), Corynebacterium (18.9%) and Virgibacillus (1.2%) (Fig. 3b). Lactobacillus was the dominant genus in PM4, sharply increasing from 1.3% in PM3 to 44.9% in PM4. The relative contents of genus Corynebacterium were 18.9% (PM4) and 16.5% (PM5), but this genus was rarely seen in the other litters. The relative abundance of Pediococcus in PM4 was also the highest among those samples, increasing from 0.1% in PM3 to 6.5% in PM4. PM5 had the highest amount of genus Bacillus (3.1%) and Virgibacillus (16.9%). The relative abundance of Pseudomonas in samples was also calculated, and the amounts of genus Pseudomonas increased to 4.5% in PM5 samples.
Relationship between bacterial communities among PM samples
A Venn diagram of the common and unique OTUs among PM libraries revealed that the number of OTUs shared among all libraries was 666 (Fig. 4). The PM1 library shared 96 and 60 OTUs with PM2 and PM3, and 66 and 44 common OTUs with PM4 and PM5. The shared OTUs accounted for >41.75% of OTUs in each group, and 3.7–8.4% of OTUs were unique in each sample.

Venn diagram based on shared OTUs in the different samples
Based on PCA supported by Weighted UniFrac distance, the five samples were divided into two significant groups in the PCA plot (Fig. 5). PM1, PM2 and PM3 grouped to the left of the plot along the first principal component (PC1), while PM4 and PM5 gathered on the right of PC1, which represented 87.24% of the total variation. PM1 and PM2 gathered together along PC2, whereas the PM3 exhibited a noticeable separation on the lower half of the graph, accounting for 6.56% of total variation.

Principal component analysis (PCA) based on OTU level among five samples
A hierarchically clustered heatmap was prepared based on the top 35 abundant bacterial communities at genus level. The heatmap plot gives an overall view of the identified connections among PM samples. As shown in Fig. 6, the five samples were classified into two groups. The first group was composed of short-term fermentation samples (PM1, PM2 and PM3). PM1 and PM2 libraries grouped firstly together, and then clustered with PM3. The other group was composed of long-term fermentation samples (PM4 and PM5).

Hierarchically clustered heatmap of bacterial distribution of different communities from the five samples at the genus level
Among the five samples, PM1 had highest relative abundance of Paludibacter (1.3%), Ruminococcus (2.5%) and TYRC22 (1.7%). Acinetobacter (8.7%), Myroides (6.4%), Sphingobacterium (0.9%) and Bacteroides (3.1%) exhibited relatively higher abundance in PM2. All those predominant strains in PM1 and PM2 were rare in long-term fermentation PM. Although PM3 shared several of the same predominant bacteria with PM1 and PM2, the dominant strains were Prevotella (2.4%), Anaerovibrio (1.1%), Dialister (0.8%), Mitsuokella (0.6%) and Megasphaera (4.6%). Sequences belonging to SMB53, Clostridium, Lactobacillus, Pediococcus, and Corynebacterium were the most dominant bacterial genus in the PM4 sample but were rare in the other samples.
The bacterial genera Pseudomonas (4.5%), Bacillus (3.1%), Virgibacillus (16.9%), Anaerococcus (3.1%), Caldicoprobacter (0.9%) and Ignatzschineria (0.9%) were rare in PM1, PM2 and PM3, but had relatively higher abundance in PM5. The abundant genus Corynebacterium was detected in both PM4 and PM5 samples. The results of the PCA and heatmap analyses indicated that the bacteria varied at different periods of PM fermentation.
RDA indicated that skatole content was the main determinant factor for the bacterial diversity of the five PM samples (Fig. 7). PM1 had high potassium and skatole content, and low C/N ratio. At the phylum level, Euryarchaeota (1.2%) and Fibrobacteres (0.6%) were the most abundant bacteria in PM1 among five samples, while the Firmicutes, Actinobacteria and Acidobacteria were predominant bacteria in PM4 and PM5. The relative abundance of Firmicutes, Actinobacteria, Acidobacteria and Proteobacteria had a close relationship with degradation of 3-MI and increasing C/N ratio.

Redundancy analysis (RDA) of the relationship between the PM physicochemical parameters and the relative abundance of bacterial phyla in the five PM samples
Discussion
Intensified and large-scale pig production leads to large amounts of manure production, and appropriate manure management is urgently needed to reduce manure emissions and environmental pollution. The MFB system has been introduced to recycle these wastes as a resource. However, research on the bacterial communities and diversity in PM in MFB systems have been lacking to date. Analysis of PM in an MFB system in situ revealed that bacterial community dynamics were related to fermentation stage. OTUs, species richness estimates and heatmap results illustrated that PM at different fermentation stages had significantly different bacterial communities. Organic degradation strains such as Bacillus, Virgibacillus, Pseudomonas, Actinobacteria and Lactobacillus might play important roles in the decomposition of PM. The amount of the unpleasant odor substance 3-MI decreased and C/N increased in the fermented manure, referred to as manure with deep fermentation in the MFB system.
Asano et al. (2010) reported that the stable dominant phylum in cattle manure was Firmicutes. In the co-composting of cow manure and rice straw, Bacteroidetes and Proteobacteria were the most abundant phyla (Ren et al. 2016). In the PM samples (PM1), a similar result was observed. The main phylum was Firmicutes, which increased from 48.7% in fresh PM to 65.5% in PM4. Since it makes up a large portion of the gut microbiome (Ley et al. 2008), Firmicutes from gut flora could be the main source of Firmicutes in PM. However, the proportion of Proteobacteria showed high variability between different samples, in the range of 7.4% to 16.8%. Lin et al. (2016) reported that most functional proteins in a thermophilic swine manure digester were from Proteobacteria. Actinobacteria was the second most abundant phylum in samples PM4 and PM5, contributing of 22.2% and 22.6% of all sequences. As Actinobacteria had extraordinary ability to decompose organic matter (Ventura et al. 2007), the increase in this phylum may have some correlation with the degradation of organic matter in PM.
As shown in the heatmap of bacterial community composition of the five samples (Fig. 4), the predominant strains changed at each stage of the fermentation process. In long-term fermentation PM, the dominant genera were related to organic degradation strains such as Corynebacterium, Bacillus, Virgibacillus, Pseudomonas, Actinobacteria, Lactobacillus and Pediococcus. Genus Corynebacterium is widely distributed in soil, water, plants, and food products, and members of genus Corynebacterium are capable of degrading aromatic compounds (Shen et al. 2012). PM5 had the highest amount of genus Bacillus (3.1%) and Virgibacillus (16.9%), which are prevalente in varied environments such as soil, bile, water and food (Heyndrickx 1998; Nicholson 2002). Bacillus spp. and Virgibacillus spp. play a key role in bio-composting processes, by producing a variety of enzymes (Hayat et al. 2013; Alkindi and Abed 2016). The relative abundances of Actinomyces and Pseudomonas in PM5 were also calculated; both demonstrated great metabolic diversity and were the most versatile and efficient hydrocarbon degraders (Folsom et al. 1990; Folsom 1997; Song et al. 2009). Liu et al. (2011) reported that Actinobacteria was the major decomposer for breaking down organic matter in composting. Prolonging compost time from 0 days to 20 days produces a sudden increase and decrease of Lactobacillus, and the appearance of Pediococcus. Lactobacillus and Pediococcus were the main lactic acid bacteria (LAB) in the five samples. LAB have been enumerated in manure and fertilized silage crops, and play important roles in composting. A LAB community comprising mainly L. fermentum, L. plantarum and L. paracacei efficiently accelerated the fermentation of straw, which could efficiently utilize rice straw and lessen its environmental disposal problems (Gao et al. 2008). The genus Pediococcus is a close relative of the genus Lactobacillus, and is used in treatment of manure. A biotechnological process in which poultry waste manure is inoculated with a starter culture of Pediococcus is a key component of composite organic waste and cattle manure fermentation systems (Asano et al. 2010; Probst et al. 2013). L. plantarum and P. acidolactici have been applied for treatment and recycling of manure as a feed ingredient (Jalil et al. 2001). In conclusion, organic degradation strains as Corynebacterium, Bacillus, Virgibacillus, Pseudomonas, Actinobacteria, Lactobacillus and Pediococcus from those samples could be involved in the degradation of PM.
LAB also represent potential 3-MI-degrading bacteria. Lactobacillus and Pediococcus of Firmicutes were the main LAB in the five samples. Meng et al. (2013) demonstrated that Lactobacillus brevis 1.12 is capable of 3-MI degradation. The large amount of LAB in PM samples might participate in the removal of skatole in PM, and be responsible for the low concentrations of 3-MI. Besides LAB, many species of Pseudomonas (phylum Proteobacteria) could also degrade 3-MI. Pseudomonas was detected in all PM samples, with relative abundance of 0.5%, 1.5%, 0.4%, 0.4% and 4.5% in PM1, PM2, PM3, PM4 and PM5, respectively. P. aeruginosa Gs isolated from Mangrove Sediment can use 3-MI as the sole source of carbon and energy (Yin et al. 2006). Cupriavidus (phylum Proteobacteria) was detected only in PM4 and PM5 samples, with quite low relative abundant of 0.004% and 0.01%, respectively. Cupriavidus sp. strain KK10 isolated from soil could transform 3-MI to ring cleavage products under aerobic conditions (Fukuoka et al. 2015). Borowski et al. (2016) investigated microbial-mineral preparation that contained Pseudomonas sp., Bacillus sp. and Lactobacillus sp. for the removal of offensive odors from poultry manure. The changes of genus Lactobacillus, Pseudomonas and Cupriavidus matched with the changes of 3-MI, and might result in the decrease of 3-MI.
This study investigated the characteristics of PM in MFB at different stages, and examined the structures of bacterial communities involved in the composting process. The diversity of bacterial communities and content of 3-MI decreased in this process. Compared with fresh PM, organic degradation strains were the dominate genera in long-term fermentation PM. Lactobacillus, Pseudomonas and Cupriavidus, as potential 3-MI-degrading bacteria, might participate in the removal of skatole in PM. This study provides data to help our understanding of bacterial metabolic activity of PM under in situ fermentation in the MFB system.
References
Alkindi S, Abed R (2016) Effect of biostimulation using sewage sludge, soybean meal, and wheat straw on oil degradation and bacterial community composition in a contaminated desert soil. Front Microbiol 7:1–14. doi:10.3389/fmicb.2016.00240
Arora PK, Shrama A, Bae H (2015) Microbial degradation of indole and its derivatives. J Chem 81:1966–1970. doi:10.1155/2015/129159
Arthurson V (2011) Storage conditions and animal source influence the dominant bacterial community composition in animal manure. World J Microbiol Biotechnol 27:2013–2022. doi:10.1007/s11274-011-0663-0
Asano R, Otawa K, Ozutsumi Y, Yamamoto N, Abdel-Mohsein HS, Nakai Y (2010) Development and analysis of microbial characteristics of an acidulo composting system for the treatment of garbage and cattle manure. J Biosci Bioeng 110:419–425. doi:10.1016/j.jbiosc.2010.04.006
Borowski S, Matusiak K, Powałowski S, Pielech-Przybylska K, Makowski K, Nowak A et al (2016) A novel microbial-mineral preparation for the removal of offensive odors from poultry manure. Int Biodeter Biodegr 119:299–308. doi:10.1016/j.ibiod.2016.10.042
Chadwick D, Jia W, Tong Y, Yu GH, Shen QR, Chen Q (2015) Improving manure nutrient management towards sustainable agricultural intensification in China. Agric Ecosyst Environ 209:34–46. doi:10.1016/j.agee.2015.03.025
Costa M, Cestonaro T, Costa L, Rozatti M, Carneiro L, Pereira D (2015) Improving the nutrient content of sheep bedding compost by adding cattle manure. J Clean Prod 86:9–14. doi:10.1016/j.jclepro.2014.08.093
Dehnhard M, BernalBarragan H, Claus R (1991) Rapid and accurate high-performance liquid chromatographic method for the determination of 3-methylindole (skatole) in faeces of various species. J Chromatogr 566:101–107. doi:10.1016/0378-4347(91)80114-R
Deslandes B, Gariepy C, Houde A (2001) Review of microbiological and biochemical effects of skatole on animal production. Livest Prod Sci 8:193–200. doi:10.1016/S0301-6226(01)00189-0
Folsom BR (1997) Characterization of 2-nitrophenol uptake system of Pseudomonas putida b2. J Ind Microbiol Biotechnol 19:123–129. doi:10.1038/sj.jim.2900432
Folsom B, Chapman P, Pritchard P (1990) Phenol and trichloroethylene degradation by Pseudomonas cepacia G4: kinetics and interactions between substrates. Appl Environ Microbiol 56:1279–1285
Fukuoka K, Ozeki Y, Kanaly RA (2015) Aerobic biotransformation of 3-methylindole to ring cleavage products by Cupriavidus sp. strain KK10. Biodegradation 26:1–15. doi:10.1007/s10532-015-9739-0
Gao L, Yang H, Wang X, Huang Z, Ishii M, Igarashi Y (2008) Rice straw fermentation using lactic acid bacteria. Bioresour Technol 99:2742–2748. doi:10.1016/j.biortech.2007.07.001
Hayat R, Sheirdil RA, Iftikhar-Ul-Hassan M (2013) Characterization and identification of compost bacteria based on 16s rRNA gene sequencing. Ann Microbiol 63:905–912. doi:10.1007/s13213-014-0889-9
Heyndrickx M (1998) Virgibacillus : a new genus to accommodate Bacillus pantothenticus (Proom and knight 1950). Amended description of Virgibacillus pantothenticus. Int J Syst Bacteriol 48:99–106. doi:10.1099/00207713-48-1-99
Jalil MH, Faid M, Elyachioui M (2001) A biotechnological process for treatment and recycling poultry wastes manure as a feed ingredient. Biomass Bioenergy 21:301–309. doi:10.1016/j.biortech.2004.12.026
Jensen MT, Cox RP, Jensen BB (1995) 3-methylindole (skatole) and indole production by mixed population of pig fecal bacteria. Appl Environ Microb 61:3180–3184
Karadag D, Özkaya B, Ölmez E, Nissilä ME, Çakmakçı M, Yıldız Ş, Puhakka JA (2013) Profiling of bacterial community in a full-scale aerobic composting plant. Int Biodeter Biodegr 77:85–90. doi:10.1016/j.ibiod.2012.10.011
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS (2008) Evolution of mammals and their gut microbes. Science 320:1647–1651. doi:10.1126/science.1155725
Li Q, Wang XC, Zhang HH, Shi HL, Hu T, Ngo HH (2013) Characteristics of nitrogen transformation and microbial community in an aerobic composting reactor under two typical temperatures. Bioresour Technol 137:270–277. doi:10.1016/j.biortech.2013.03.092
Lin YW, Tuan NN, Huang SL (2016) Metaproteomic analysis of the microbial community present in a thermophilic swine manure digester to allow functional characterization: a case study. Int Biodeter Biodegr 115:64–73 http://dx.doi.org/10.1016/j.ibiod.2016.06.013
Liu D, Zhang R, Wu H, Xu D, Tang Z, Yu G (2011) Changes in biochemical and microbiological parameters during the period of rapid composting of dairy manure with rice chaff. Bioresour Technol 102:9040–9049. doi:10.1016/j.biortech.2011.07.052
López-González JA, Suárez-Estrella F, Vargas-García MC, López MJ, Jurado MM, Moreno J (2014) Dynamics of bacterial microbiota during lignocellulosic waste composting: studies upon its structure, functionality and biodiversity. Bioresour Technol 175:406–416. doi:10.1016/j.biortech.2014.10.123
Menalled FD, Buhler DD, Liebman M (2005) Composted swine manure effects on germination and early growth of crop and weed species under greenhouse conditions. Weed Technol 19:784–789. doi:10.1614/WT-04-224.1
Meng X, He ZF, Li HJ, Zhao X (2013) Removal of 3-methylindole by lactic acid bacteria in vitro. Exp Ther Med 6:983–988. doi:10.3892/etm.2013.1251
Morrison RS, Johnston LJ, Hilbrands AM (2007) The behavior, welfare, growth performance and meat quality of pigs housed in a deep-litter, large group housing system compared to a conventional confinement system. Appl Anim Behav Sci 103:12–24. doi:10.1016/j.applanim.2006.04.002
Nakai Y, Niino T, Ando T, Kohda C (1999) Microorganisms aerobically degrading skatole or indole in composting processes. Nihon Chikusa Gakkaiho 70:32–37. doi:10.2508/chikusan.70.32
Neumann EJ, Kliebenstein JB, Johnson CD, Mabry JW, Bush EJ, Seitzinger AH (2015) Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the US. J Am Vet Med A 227:385–392. doi:10.2460/javma.2005.227.385
Ni JQ, Robarge WP, Xiao C, Heber AJ (2012) Volatile organic compounds at swine facilities: a critical review. Chemosphere 89:769–788. doi:10.1016/j.chemosphere.2012.04.061
Nicholson WL (2002) Roles of Bacillus endospores in the environment. Cell Mol Life Sci 59:410–416. doi:10.1007/s00018-002-8433-7
Probst M, Fritschi A, Wagner A, Insam H (2013) Biowaste: A. Lactobacillus, habitat and lactic acid fermentation substrate. Bioresour Technol 143:647–652. doi:10.1016/j.biortech.2013.06.022
Rashad FM, Saleh WD, Moselhy MA (2010) Bioconversion of rice straw and certain agro-industrial wastes to amendments for organic farming systems: composting, quality, stability and maturity indices. Bioresour Technol 101:5952–5960. doi:10.1016/j.biortech.2010.02.103
Ren G, Xu X, Qu J, Zhu L, Wang T (2016) Evaluation of microbial population dynamics in the co-composting of cow manure and rice straw using high throughput sequencing analysis. World J Microb Biot 32:1–11. doi:10.1007/s11274-016-2059-7
Shen X, Zhou N, Liu S (2012) Degradation and assimilation of aromatic compounds by Corynebacterium glutamicum: another potential for applications for this bacterium. Appl Microbiol Biotechnol 95:77–89. doi:10.1007/s00253-012-4139-4
Song H, Liu Y, Xu W, Zeng G, Aibibu N, Xu L (2009) Simultaneous Cr(VI) reduction and phenol degradation in pure cultures of Pseudomonas Aeruginosa CCTCC AB91095. Bioresour Technol 100:5079–5084. doi:10.1016/j.biortech.2009.05.060
Tam NFY (1995) Changes in microbiological properties during in-situ composting of pig manure. Environ Technol 16:445–456. doi:10.1080/09593331608616285
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, Sinderen D (2007) Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol R 71:495–548. doi:10.1128/MMBR.00005-07
Yin B, Huang L, Gu JD (2006) Biodegradation of 1-methylindole and 3-methylindole by mangrove sediment enrichment cultures and a pure culture of an isolated Pseudomonas aeruginosa Gs. Water Air Soil Poll 176:185–199. doi:10.1007/s11270-006-9159-1
Zheng X, Liu B, Lan J, Su M, Lu S, Zhu C (2011) Study on the biocontrol effects of microbial fermentation bed on the pig pathogen Escherichia coil in the piggery. Sci Agric Sin 44:4728–4739. doi:10.3864/j.issn.0578-1752.2011.22.022
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (grant #31370059); the Fujian Key Science and Technology Special Projects—Key Agricultural Science and Technology Special Project (grant #2015NZ0003-1).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, Q., Liu, B., Wang, J. et al. Diversity and dynamics of the bacterial community involved in pig manure biodegradation in a microbial fermentation bed system. Ann Microbiol 67, 491–500 (2017). https://doi.org/10.1007/s13213-017-1278-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13213-017-1278-y